
38 系統樹
38.1 概要
系統樹とは、生物の遺伝暗号の配列に基づいて、その近縁性や進化を可視化・記述するためのものです。
系統樹は、遺伝子配列から、距離に基づく方法(近傍結合法など)や、特徴に基づく方法(最尤法やベイズ・マルコフ連鎖モンテカルロ法など)を用いて構築することができます。次世代シーケンシング(NGS)は、価格が手頃になり、感染症の原因となる病原体を記述するために公衆衛生の分野で広く使われるようになってきました。携帯型のシーケンサーを使用することで、解析時間を短縮し、リアルタイムでアウトブレイク調査に必要なデータを得ることができます。NGS データは、発生した菌株の起源や発生源、その繁殖を特定したり、抗菌剤耐性遺伝子の有無を判定したりするのに利用できます。また、サンプル間の遺伝的関連性を可視化するために、系統樹を構築します。
このページでは、系統樹とデータフレーム形式の追加サンプルデータを組み合わせて可視化することができる ggtree パッケージの使い方を学びます。これにより、パターンを観察し、アウトブレイクのダイナミックな理解を深めることができます。
38.2 準備
パッケージの読み込み
このコードチャンクは、分析に必要なパッケージの読み込みを示しています。このハンドブックでは pacman の p_load() を重視しています。p_load() は必要に応じてパッケージをインストールし、使用するためにパッケージを読み込みます。インストールされたパッケージは R の base パッケージの library() でも読み込みできます。R のパッケージに関する詳細は R の基礎 の章をご覧ください。
pacman::p_load(
rio, # インポート/エクスポート
here, # 相対的なファイルパス
tidyverse, # 一般的なデータマネジメントと可視化
ape, # 系統樹ファイルのインポートとエクスポート
ggtree, # 系統樹の可視化
treeio, # 系統樹の可視化
ggnewscale) # 色に関するレイヤーの追加データのインポート
本章で扱われるデータは、ハンドブックとデータのダウンロード の章の説明に従ってダウンロードできます。
系統樹の保存形式にはいくつかの種類があります( Newick、NEXUS、Phylip など)。一般的なのはNewickファイル形式(.nwk)で、これは木をコンピュータで読める形で表現するための標準的なものです。つまり、木全体を “((t2:0.04,t1:0.34):0.89,(t5:0.37,(t4:0.03,t3:0.67):0.9):0.59);” のような文字列形式で表現し、すべてのノードと先端、そしてそれらの関係(枝の長さ)を列挙することができます。
注: 系統樹ファイル自体にはシーケンスデータは含まれておらず、単に配列間の遺伝的距離の結果であることを理解しておくことが重要です。したがって、ツリーファイルからシーケンスデータを抽出することはできません。
まず、ape パッケージの read.tree() 関数を使って、Newick の系統樹ファイルを .txt 形式で読み込み、“phylo” 型のリストオブジェクトに格納しておきます。必要に応じて、here パッケージの here() 関数を使用して、相対ファイルパスを指定します。
注:このケースでは、Github からのダウンロードや取り扱いを容易にするために、newick ツリーを .txt ファイルとして保存しています。
tree <- ape::read.tree("Shigella_tree.txt")tree オブジェクトを調べてみると、299 個の先端(またはサンプル)と 236 個のノードが含まれています。
tree
Phylogenetic tree with 299 tips and 236 internal nodes.
Tip labels:
SRR5006072, SRR4192106, S18BD07865, S18BD00489, S17BD08906, S17BD05939, ...
Node labels:
17, 29, 100, 67, 100, 100, ...
Rooted; includes branch lengths.次に、rio パッケージの import() 関数を用いて、性別、原産国、抗菌薬への薬剤耐性など、シーケンスされた各サンプルの追加情報を含む .csv ファイルとして保存された表をインポートします。
sample_data <- import("sample_data_Shigella_tree.csv")以下は、データの最初の 50 行です。
クリーニングと点検
データのクリーニングと点検を行います。正しいサンプルデータを系統樹に割り当てるためには、sample_data データフレームの Sample_ID カラムの値が、tree ファイルの tip.labels の値と一致する必要があります。
R の base パッケージの head() を使って最初の 6 つのエントリーを見ることで、 tree ファイルの tip.labels のフォーマットをチェックします。
head(tree$tip.label) [1] "SRR5006072" "SRR4192106" "S18BD07865" "S18BD00489" "S17BD08906"
[6] "S17BD05939"また、sample_data データフレームの最初の列が Sample_ID であることを確認します。R の base パッケージの colnames() を使って、データフレームのカラム名を調べます。
colnames(sample_data) [1] "Sample_ID" "serotype"
[3] "Country" "Continent"
[5] "Travel_history" "Year"
[7] "Belgium" "Source"
[9] "Gender" "gyrA_mutations"
[11] "macrolide_resistance_genes" "MIC_AZM"
[13] "MIC_CIP" データフレーム内の Sample_IDs を見て、フォーマットが tip.label と同じであることを確認します(例:文字はすべて大文字、文字と数字の間に余分なアンダースコア _ がない、など)。
head(sample_data$Sample_ID) # もう一度、head() を使用して、初めの 6 つのデータだけ確認する[1] "S17BD05944" "S15BD07413" "S18BD07247" "S19BD07384" "S18BD07338"
[6] "S18BD02657"また、すべてのサンプルが tree ファイルに存在するかどうか、あるいはその逆かどうかを、一致する部分と一致しない部分に TRUE または FALSE のロジカルベクトルを生成することで比較することができます。これらは、簡単にするために、ここでは表示しません。
sample_data$Sample_ID %in% tree$tip.label
tree$tip.label %in% sample_data$Sample_IDこれらのベクトルを使って、 tree 上にないサンプル ID を表示することができます(結果として、そのような例はありません)。
sample_data$Sample_ID[!tree$tip.label %in% sample_data$Sample_ID]character(0)調べてみると、データフレーム内の Sample_ID のフォーマットが、tip.labels のサンプル名のフォーマットに対応していることがわかります。これらは、同じ順序でソートされていなくてもマッチします。
これで準備完了です。
38.3 単純な系統樹の可視化
様々な系統樹のレイアウト
ggtree には様々なレイアウトが用意されており、目的に応じて最適なものを選ぶことができます。以下にいくつかのデモンストレーションを紹介します。他のオプションについては、このオンラインブックを参照してください。
以下は、ツリーレイアウトの例です。
ggtree(tree) # 単純な線形の系統樹
ggtree(tree, branch.length = "none") # 全ての先端が揃えられた単純な線形の系統樹
ggtree(tree, layout="circular") # 単純な円形の系統樹
ggtree(tree, layout="circular", branch.length = "none") # 全ての先端が揃えられた単純な円形の系統樹



単純な系統樹とサンプルデータ
sample_data データフレームを tree ファイルに接続するには、%<+% 演算子を使用します。 系統樹の最も簡単な注釈は、先端にサンプル名を追加することと、先端のポイントや必要に応じて枝に色を付けることです。
以下は、円形の系統樹の例です。
ggtree(tree, layout = "circular", branch.length = 'none') %<+% sample_data + # %<+% で、tree にサンプルデータを追加する
aes(color = Belgium)+ # データフレームの変数に応じて分枝に色付ける
scale_color_manual(
name = "Sample Origin", # カラースキームの名前
breaks = c("Yes", "No"), # 変数の異なるオプション
labels = c("NRCSS Belgium", "Other"), # 凡例の中で異なるオプションをどのように命名するか
values = c("blue", "black"), # 変数に割り当てたい色
na.value = "black") + # NA 値を黒に
new_scale_color()+ # 別の変数の配色を追加する
geom_tippoint(
mapping = aes(color = Continent), # 大陸別の先端の色。"shape = " を加えて形状を変更する
size = 1.5)+ # 先端のポイントのサイズを定義する
scale_color_brewer(
name = "Continent", # カラースキームの名前(凡例ではこのように表示される
palette = "Set1", # brewer パッケージに付属しているカラーセットを選ぶ
na.value = "grey") + # NA 値はグレーを選択
geom_tiplab( # 枝の先端にサンプルの名前を追加
color = 'black', # (テキストラインは + で好きなだけ追加できるが、隣り合うように配置するにはオフセット値を調整する必要がある)
offset = 1,
size = 1,
geom = "text",
align = TRUE)+
ggtitle("Phylogenetic tree of Shigella sonnei")+ # グラフのタイトル
theme(
axis.title.x = element_blank(), # x 軸のタイトルを削除
axis.title.y = element_blank(), # y 軸のタイトルを削除
legend.title = element_text( # 凡例のタイトルのフォントサイズとフォーマットを定義する
face = "bold",
size = 12),
legend.text=element_text( # 凡例テキストのフォントサイズとフォーマットを定義する
face = "bold",
size = 10),
plot.title = element_text( # プロットタイトルのフォントサイズとフォーマットを定義する
size = 12,
face = "bold"),
legend.position = "bottom", # 凡例の配置を決める
legend.box = "vertical", # 凡例の配置を決める
legend.margin = margin()) 
他の ggplot オブジェクトと同じように、ggsave() を使ってツリープロットをエクスポートすることができます。このように書くと、ggsave() は最後に生成された画像を指定されたファイルパスに保存します。here() や相対ファイルパスを使えば、サブフォルダなどにも簡単に保存できることを覚えておいてください。
ggsave("example_tree_circular_1.png", width = 12, height = 14)38.4 系統樹の操作・処理・加工
非常に大きな系統樹であっても、その中の一部分にしか興味がない場合があります。例えば、歴史的または国際的なサンプルを含む系統樹を作成して、データセットが全体像の中でどこに当てはまるかを大まかに把握する場合です。しかし、データをより詳しく見るためには、大きな系統樹の一部だけを確認したいとします。
系統樹ファイルはシーケンシングデータの解析結果に過ぎないので、ファイル内のノードや枝の順番を操作することはできません。これらは、生の NGS データから以前の解析ですでに決定されています。しかし、部分的に拡大したり、部分的に非表示にしたり、系統樹の一部をサブセットにしたりすることは可能です。
拡大
系統樹を “切断” するのではなく、一部だけをより詳しく調べたい場合は、ズームインして特定の部分を表示することができます。
まず、系統樹全体を線形のフォーマットでプロットし、系統樹の各ノードに数字のラベルを追加します。
p <- ggtree(tree,) %<+% sample_data +
geom_tiplab(size = 1.5) + # すべての枝の先端に、tree ファイルのサンプル名をラベル付ける
geom_text2(
mapping = aes(subset = !isTip,
label = node),
size = 5,
color = "darkred",
hjust = 1,
vjust = 1) # 系統樹内のすべてのノードにラベルを付ける
p # 表示する
ある特定の枝(右に突き出ている)にズームインするには、ggtree オブジェクト p で viewClade() を使用し、ノード番号を指定すると、より詳しく見ることができます。
viewClade(p, node = 452)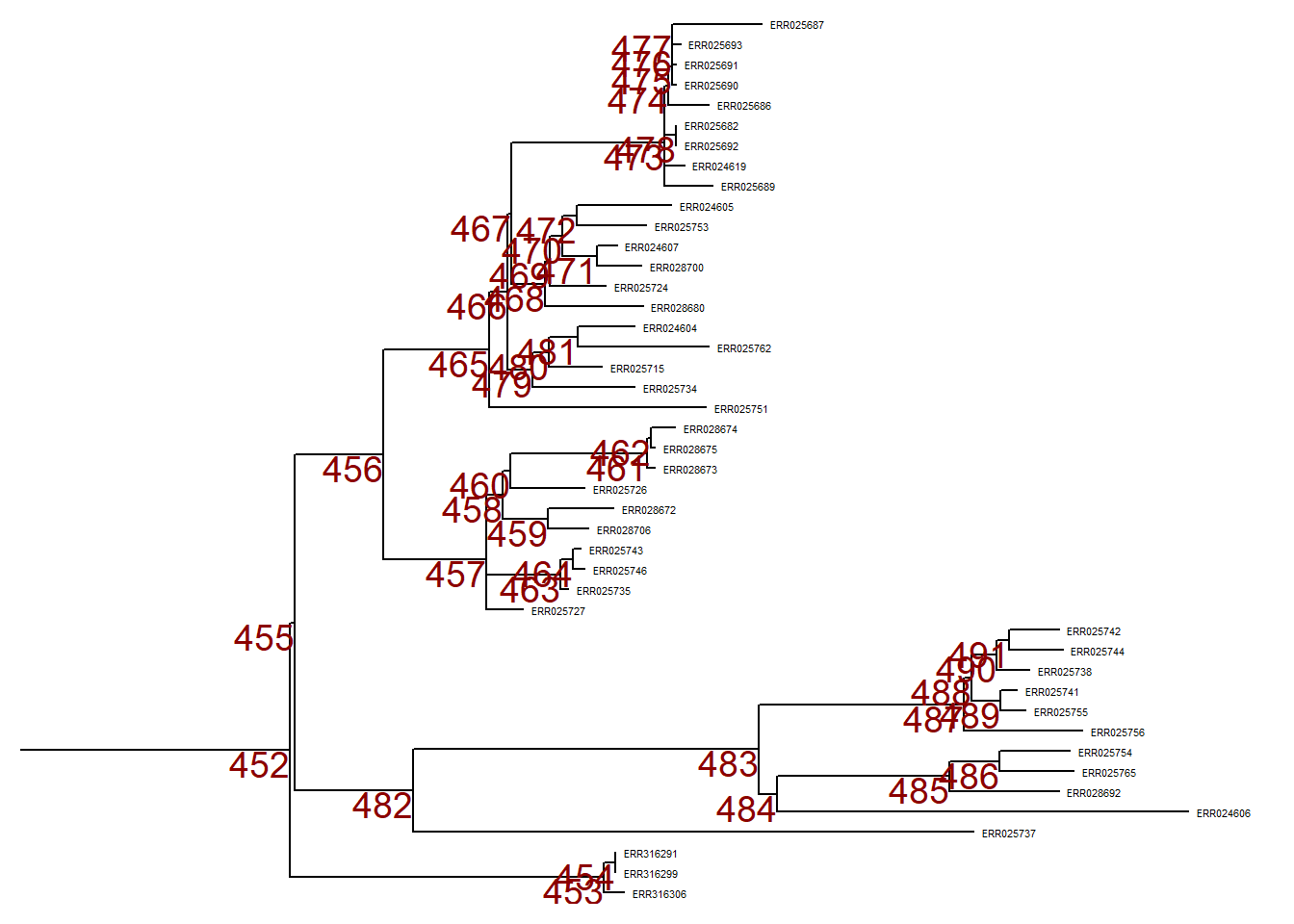
枝の折り畳み
しかし、この枝を無視したい場合もあるので、collapse() を使って同じノード(ノード番号 452)で枝を折り畳むことができます。このツリーは p_collapsed として定義されます。
p_collapsed <- collapse(p, node = 452)
p_collapsed
分かりやすくするために、p_collapsed をプリントする際に、折り畳まれた枝のノードに geom_point2()(青い菱形)を追加しています。
p_collapsed +
geom_point2(aes(subset = (node == 452)), # 折り畳んだノードに記号を割り当てる
size = 5, # 記号のサイズを定義する
shape = 23, # 記号の形を定義する
fill = "steelblue") # 記号の塗りつぶしを定義する
系統樹の分割(サブセット)
より永続的な変更を行い、作業用の縮小された新しい系統樹を作成したい場合は、 tree_subset() でツリーの一部をサブセットします。そして、それを新しい newick tree ファイルか .txt ファイルとして保存します。
ggtree(
tree,
branch.length = 'none',
layout = 'circular') %<+% sample_data + # %<+% 演算子を使って、サンプルデータを追加する
geom_tiplab(size = 1)+ # すべての枝の先端にサンプル名をラベル付けしてツリーファイルに保存
geom_text2(
mapping = aes(subset = !isTip, label = node),
size = 3,
color = "darkred") + # 系統樹内のすべてのノードにラベルを付ける
theme(
legend.position = "none", # 凡例も全て削除
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(size = 12, face="bold"))
ここで、ノード 528 で系統樹をサブセットする(ノード 528 以降の枝の中の先端だけを残す)ことにして、それを新しい sub_tree1 オブジェクトとして保存したとします。
sub_tree1 <- tidytree::tree_subset(
tree,
node = 528) # ノード 528 で系統樹をサブセットするsubset tree 1 を見てみましょう。
ggtree(sub_tree1) +
geom_tiplab(size = 3) +
ggtitle("Subset tree 1")
また、1 つの特定のサンプルに基づいてサブセットすることもできます。その際には、含める「後ろ」のノード数を指定します。系統樹の同じ部分を、サンプル(ここでは S17BD07692)に基づいて 9 ノードさかのぼってサブセットし、新しい sub_tree2 オブジェクトとして保存します。
sub_tree2 <- tidytree::tree_subset(
tree,
"S17BD07692",
levels_back = 9) # levels back は、先端(各サンプル)から何ノード後方に移動するかを定義するsubset tree 2 を見てみましょう。
ggtree(sub_tree2) +
geom_tiplab(size =3) +
ggtitle("Subset tree 2")
また、ape パッケージの write.tree() 関数を使って、新しい系統樹を Newick 型やテキストファイルとして保存することもできます。
# .nwk フォーマットで保存
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.nwk')
# .txt フォーマットで保存
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.txt')系統樹内でのノードの回転
前述したように、ツリー内の先端やノードの順序を変更することはできません。これは、遺伝子の関連性に基づいており、視覚的な操作の対象にはならないからです。しかし、視覚的にわかりやすくするために、ノードの周りに枝を回転させることはできます。
まず、ノードのラベルとともに新しい subset tree 2 をプロットして、操作したいノードを選び、それを ggtree plot オブジェクト p に格納します。
p <- ggtree(sub_tree2) +
geom_tiplab(size = 4) +
geom_text2(aes(subset=!isTip, label=node), # 系統樹内のすべてのノードにラベルを付ける
size = 5,
color = "darkred",
hjust = 1,
vjust = 1)
p
その後、 ggtree::rotate() や ggtree::flip() を使ってノードを操作することができます。 注:どのノードを操作しているかを示すために、まず、ggtree の geom_hilight() 関数を適用して、興味のあるノードのサンプルをハイライトし、その ggtree プロットオブジェクトを新しいオブジェクト p1 に格納します。
p1 <- p + geom_hilight( # ノード 39 を青くハイライトし、"extend =" でカラーブロックの長さを定義する
node = 39,
fill = "steelblue",
extend = 0.0017) +
geom_hilight( # ノード 37 を黄色くハイライトする
node = 37,
fill = "yellow",
extend = 0.0017) +
ggtitle("Original tree")
p1 # 表示する
ここで、オブジェクト p1 のノード 37 を回転させ、ノード 38 上のサンプルが一番上に移動するようにします。回転したツリーを新しいオブジェクト p2 に格納します。
p2 <- ggtree::rotate(p1, 37) +
ggtitle("Rotated Node 37")
p2 # 表示する
また、flip コマンドを使って、オブジェクト p1 のノード 36 を回転させ、ノード 37 を上に、ノード 39 を下に切り替えることもできます。反転したツリーを新しいオブジェクト p3 に格納します。
p3 <- flip(p1, 39, 37) +
ggtitle("Rotated Node 36")
p3 # 表示する
サンプルデータを付加したサブツリーの例
サブツリーのノード 39 で 2017 年と 2018 年に発生したクローン拡大を伴う症例のクラスタを調査しているとします。他の近縁種の株の起源を見るために、株の分離年に加えて、渡航歴と国別の色を追加します。
ggtree(sub_tree2) %<+% sample_data + # %<+% 演算子を使って sample_data にリンクしている
geom_tiplab( # すべての枝の先端に、ツリーファイルのサンプル名をラベル付けする
size = 2.5,
offset = 0.001,
align = TRUE) +
theme_tree2()+
xlim(0, 0.015)+ # 系統樹の x 軸の 下限、上限を設定
geom_tippoint(aes(color=Country), # 大陸別に先端を色付ける
size = 1.5)+
scale_color_brewer(
name = "Country",
palette = "Set1",
na.value = "grey")+
geom_tiplab( # 隔離された年をテキストラベルとして先端に追加
aes(label = Year),
color = 'blue',
offset = 0.0045,
size = 3,
linetype = "blank" ,
geom = "text",
align = TRUE)+
geom_tiplab( # 先端のテキストラベルに渡航歴を赤で表示
aes(label = Travel_history),
color = 'red',
offset = 0.006,
size = 3,
linetype = "blank",
geom = "text",
align = TRUE)+
ggtitle("Phylogenetic tree of Belgian S. sonnei strains with travel history")+ # プロットタイトルの追加
xlab("genetic distance (0.001 = 4 nucleotides difference)")+ # x 軸にラベルを付ける
theme(
axis.title.x = element_text(size = 10),
axis.title.y = element_blank(),
legend.title = element_text(face = "bold", size = 12),
legend.text = element_text(face = "bold", size = 10),
plot.title = element_text(size = 12, face = "bold"))
私たちの観察によると、アジアからの菌株の輸入があり、それが何年もかけてベルギーで伝播し、今回の流行を引き起こしたと考えられます。
より複雑な系統樹:サンプルデータのヒートマップの追加
ggtree::gheatmap() 関数を使用して、抗菌薬耐性遺伝子のカテゴリー的な存在や、実際に測定された抗菌薬耐性の数値など、より複雑な情報をヒートマップの形で追加することができます。
まず、系統樹をプロットし(これは線形でも円形でも構いません)、それを新しい ggtree plot のオブジェクト p に保存する必要があります。ここでは、パート 3 の sub_tree を使用します)。
p <- ggtree(sub_tree2, branch.length='none', layout='circular') %<+% sample_data +
geom_tiplab(size =3) +
theme(
legend.position = "none",
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(
size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p
次に、データの準備です。異なる変数を新しい配色で可視化するために、データフレームを目的の変数にサブセットします。行の名前として Sample_ID を追加することが重要で、そうしないとデータを tip.labels の系統樹にマッチさせることができません。
この例では、性別と、赤痢菌感染症の治療に使用される重要な第一選択抗菌薬であるシプロフロキサシンへの耐性を与える可能性のある変異を調べたいと思います。
性別のデータフレームを作成します。
gender <- data.frame("gender" = sample_data[,c("Gender")])
rownames(gender) <- sample_data$Sample_IDシプロフロキサシン耐性をもたらす gyrA 遺伝子の変異について、データフレームを作成します。
cipR <- data.frame("cipR" = sample_data[,c("gyrA_mutations")])
rownames(cipR) <- sample_data$Sample_ID実験室で測定されたシプロフロキサシンの最小発育阻止濃度(MIC)のデータフレームを作成します。
MIC_Cip <- data.frame("mic_cip" = sample_data[,c("MIC_CIP")])
rownames(MIC_Cip) <- sample_data$Sample_ID系統樹に性別の二値のヒートマップを追加した最初のプロットを作成し、それを新しい ggtree plot のオブジェクト h1 に格納します。
h1 <- gheatmap(p, gender, # 性別データフレームのヒートマップレイヤーをツリープロットに追加する
offset = 10, # offset は、ヒートマップを右方向に移動する
width = 0.10, # width は、ヒートマップのカラムの幅を定義する
color = NULL, # color は、ヒートマップの列の境界線を定義する
colnames = FALSE) + # ヒートマップのカラム名を隠す
scale_fill_manual(name = "Gender", # 性別の配色と凡例の定義
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h1
その後、シプロフロキサシンに対する耐性を付与する gyrA 遺伝子の変異の情報を追加しています。
注:WGS データにおける染色体の点変異の有無は、Zankari らが開発した PointFinder ツールを用いて事前に判断しています(参考文献の項を参照)。
まず、既存のプロットオブジェクト h1 に新しい配色を割り当てて、今のオブジェクト h2 に格納します。これにより、ヒートマップの 2 つ目の変数の色を定義・変更することができます。
h2 <- h1 + new_scale_fill() 次に、2 つ目のヒートマップレイヤーを h2 に追加し、合成したプロットを新しいオブジェクト h3 に格納します。
h3 <- gheatmap(h2, cipR, # ヒートマップの 2 行目に Ciprofloxacin の耐性変異を追加
offset = 12,
width = 0.10,
colnames = FALSE) +
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h3
以上のプロセスを繰り返し、まず既存のオブジェクト h3 に新しいカラースケール層を追加し、次に得られたオブジェクト h4 に各菌株に対するシプロフロキサシンの最小発育阻止濃度(MIC)の連続データを追加して、最終的なオブジェクト h5 を作成します。
# まず、新しいカラーリングを追加する
h4 <- h3 + new_scale_fill()
# そして、その 2 つを組み合わせて新しいプロットを作る
h5 <- gheatmap(h4, MIC_Cip,
offset = 14,
width = 0.10,
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin", # ここでは、MIC の連続変数のグラデーションカラースキームを定義する
low = "yellow", high = "red",
breaks = c(0, 0.50, 1.00),
na.value = "white") +
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h5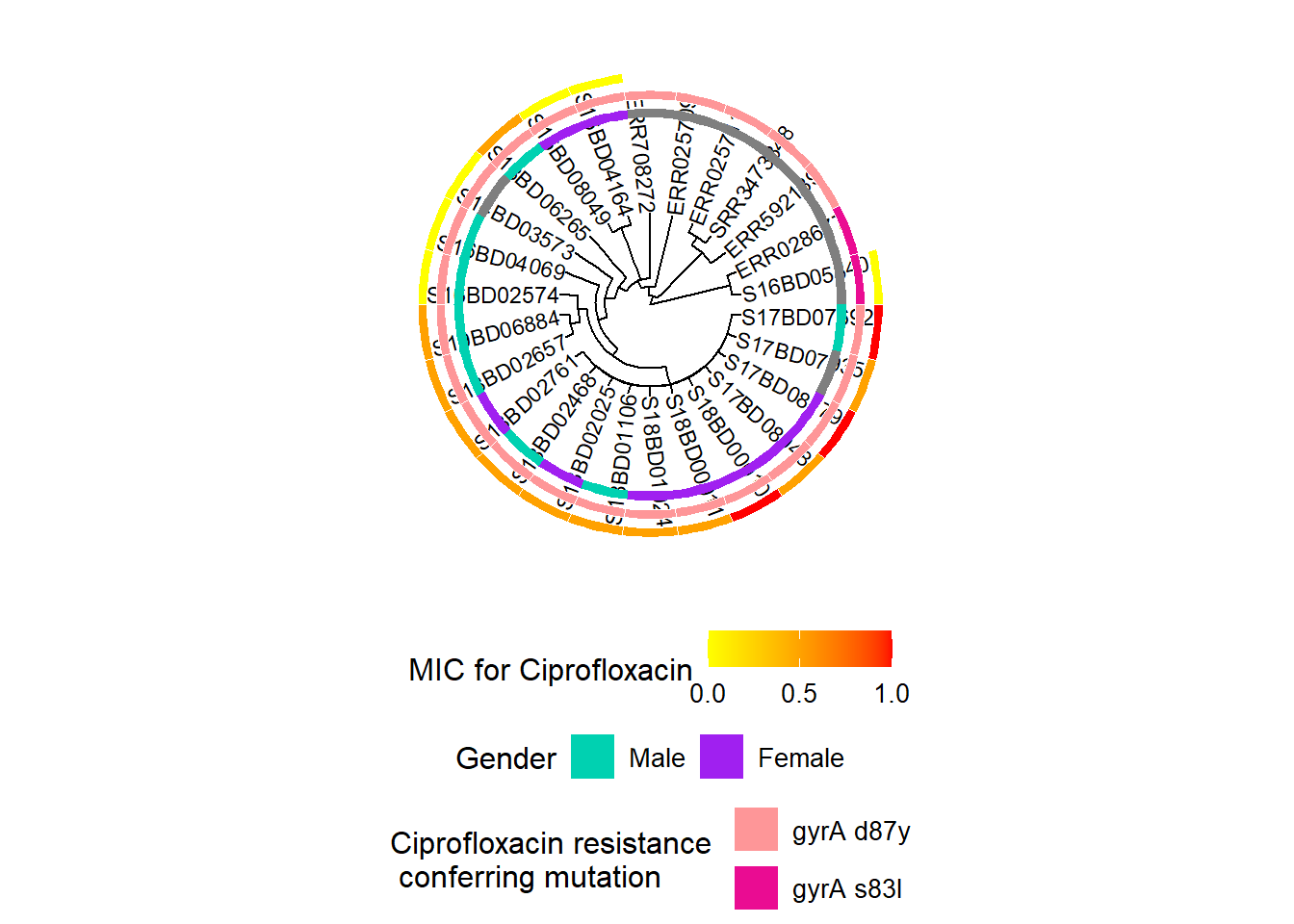
これと同じことを、線形の系統樹についても行うことができます。
p <- ggtree(sub_tree2) %<+% sample_data +
geom_tiplab(size = 3) + # 先端にラベル付け
theme_tree2()+
xlab("genetic distance (0.001 = 4 nucleotides difference)")+
xlim(0, 0.015)+
theme(legend.position = "none",
axis.title.y = element_blank(),
plot.title = element_text(size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p
まず、性別を追加します。
h1 <- gheatmap(p, gender,
offset = 0.003,
width = 0.1,
color="black",
colnames = FALSE)+
scale_fill_manual(name = "Gender",
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h1
そして、もう一つの配色レイヤーを追加した後に、シプロフロキサシン耐性変異を追加します。
h2 <- h1 + new_scale_fill()
h3 <- gheatmap(h2, cipR,
offset = 0.004,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale. h3
そして、実験室で決められた最小発育阻止濃度(MIC)を加えます。
h4 <- h3 + new_scale_fill()
h5 <- gheatmap(h4, MIC_Cip,
offset = 0.005,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin",
low = "yellow", high = "red",
breaks = c(0,0.50,1.00),
na.value = "white")+
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 10),
legend.text = element_text(size = 8),
legend.box = "horizontal", legend.margin = margin())+
guides(shape = guide_legend(override.aes = list(size = 2)))Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
Scale for fill is already present.
Adding another scale for fill, which will replace the existing scale.h5
38.5 参考資料
- http://hydrodictyon.eeb.uconn.edu/eebedia/index.php/Ggtree# Clade_Colors
- https://bioconductor.riken.jp/packages/3.2/bioc/vignettes/ggtree/inst/doc/treeManipulation.html
- https://guangchuangyu.github.io/ggtree-book/chapter-ggtree.html
- https://bioconductor.riken.jp/packages/3.8/bioc/vignettes/ggtree/inst/doc/treeManipulation.html
Ea Zankari, Rosa Allesøe, Katrine G Joensen, Lina M Cavaco, Ole Lund, Frank M Aarestrup, PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens, Journal of Antimicrobial Chemotherapy, Volume 72, Issue 10, October 2017, Pages 2764–2768, https://doi.org/10.1093/jac/dkx217