38 Filogenetik ağaçlar
38.1 Genel Bakış
Filogenetik ağaçlar, organizmaların genetik kodlarının sekansına göre akrabalıklarını ve evrimlerini görselleştirmek ve tanımlamak için kullanılır. Uzaklığa dayalı yöntemler (komşu birleştirme yöntemi gibi) veya karakter tabanlı yöntemler (maksimum olabilirlik ve Bayesian Markov Zinciri Monte Carlo yöntemi gibi) kullanılarak genetik sekanslardan oluşturulabilirler. Yeni nesil sekanslama (NGS) daha uygun fiyatlı hale gelerek bulaşıcı hastalıklara neden olan patojenleri tanımlamak için halk sağlığında daha yaygın olarak kullanılmaya başlandı. Taşınabilir sekanslama cihazları, bekleme süresini azaltır ve verileri gerçek zamanlı olarak salgın araştırmasını desteklemek için kullanılabilir hale getirir. NGS verileri, bir salgın suşunun kökenini, kaynağını ve yayılımını belirlemek ve ayrıca antimikrobiyal direnç genlerinin varlığını göstermek için kullanılabilir. Örnekler arasındaki genetik ilişkiyi görselleştirmek için bir filogenetik ağaç oluşturulur. Bu sayfada, filogenetik ağaçların bir veri çerçevesiyle birleştirilmiş halde görselleştirilmesine izin veren ggtree paketinin nasıl kullanılacağını öğreneceğiz. Bu, motifleri gözlemlememizi ve salgın dinamiğinin anlaşılmasını geliştirmemizi sağlayacaktır.
38.2 Hazırlık
Paketlerin yüklenmesi
Bu kod parçası, gerekli paketlerin yüklenmesini göstermektedir. Bu el kitabında, gerekirse paketi kuran ve kullanım için yükleyen pacman’ın p_load() fonksiyonunu vurguluyoruz. Ayrıca, temel R’dan library() ile kurulu paketleri yükleyebilirsiniz. R paketleri hakkında daha fazla bilgi için R’ın temelleri sayfasına bakın.
pacman::p_load(
rio, # içe/dışa aktarım
here, # ilişkili dosya yolları
tidyverse, # genel veri yönetimi ve görselleştirme
ape, # filogenetik dosyaları içe ve dışa aktarmak için
ggtree, # filogenetik dosyaları görselleştirmek için
treeio, # filogenetik dosyaları görselleştirmek için
ggnewscale) # ek renk şema katmanları eklemek içinVerilerin içe aktarımı
Bu sayfadaki veriler, El kitabı ve verilerin indirilmesi sayfasındaki talimatlarla indirilebilir.
Bir filogenetik ağacın saklanabileceği birkaç farklı format vardır (örn. Newick, NEXUS, Phylip). Yaygın olanlardan biri, ağaçların bilgisayar tarafından okunabilir biçimde gösterimi için standart olan Newic dosya biçimidir (.nwk).
Bu, tüm ağacın “((t2:0.04,t1:0.34):0.89,(t5:0.37,(t4:0.03,t3:0.67):0.9):0.59);” gibi bir dize biçiminde ifade edilebileceği anlamına gelir. Bu dize tüm nodları ve uçları ve bunların birbirleriyle olan ilişkilerini (dal uzunluğu) listeler.
Not: Filogenetik ağaç dosyasının kendi içinde sekans verileri içermediğini, yalnızca sekanslar arasındaki genetik mesafelerin bir sonucu olduğunu anlamak önemlidir. Bu nedenle, bir ağaç dosyasından sekanslama verilerini çıkaramıyoruz.
İlk olarak, bir Newick filogenetik ağaç dosyasını .txt formatında içe aktarmak için ape paketinden read.tree() fonksiyonunu kullanırız ve “phylo” sınıfı bir liste nesnesinde saklarız. Gerekirse, ilgili dosya yolunu belirtmek için here paketindeki here() fonksiyonunu kullanın.
Not: Bu durumda newick ağacı, Github’dan daha kolay kullanım ve indirme için bir .txt dosyası olarak kaydedilir.
tree <- ape::read.tree("Shigella_tree.txt")Ağaç nesnemizi inceliyoruz ve 299 uç (veya örnek) ve 236 nod içerdiğini görüyoruz.
tree##
## Phylogenetic tree with 299 tips and 236 internal nodes.
##
## Tip labels:
## SRR5006072, SRR4192106, S18BD07865, S18BD00489, S17BD08906, S17BD05939, ...
## Node labels:
## 17, 29, 100, 67, 100, 100, ...
##
## Rooted; includes branch lengths.İkinci olarak, rio paketindeki import() fonksiyonunu kullanarak cinsiyet, menşe ülke ve antimikrobiyal direnç özellikleri gibi her numune için ek bilgiler içeren bir .csv dosyası olarak saklanan tabloyu içe aktarırız:
sample_data <- import("sample_data_Shigella_tree.csv")Aşağıda verilerin ilk 50 satırı verilmiştir:
Temizleme ve inceleme
Verilerimizi temizler ve inceleriz: Filogenetik ağaca doğru örnek verilerini atamak için, sample_data veri çerçevesindeki Sample_ID sütunundaki değerlerin ağaç dosyasındaki tip.labels değerleriyle eşleşmesi gerekir:
Ağaç dosyasındaki tip.labels formatını, R tabanından head() ile ilk 6 girişe bakarak kontrol ederiz.
head(tree$tip.label) ## [1] "SRR5006072" "SRR4192106" "S18BD07865" "S18BD00489" "S17BD08906" "S17BD05939"Ayrıca sample_data veri çerçevemizdeki ilk sütunun Sample_ID olduğundan emin oluruz. Temel R’dan colnames() kullanarak veri çerçevemizin sütun adlarına bakarız.
colnames(sample_data) ## [1] "Sample_ID" "serotype"
## [3] "Country" "Continent"
## [5] "Travel_history" "Year"
## [7] "Belgium" "Source"
## [9] "Gender" "gyrA_mutations"
## [11] "macrolide_resistance_genes" "MIC_AZM"
## [13] "MIC_CIP"Biçimlendirmenin tip.label’dekiyle aynı olduğundan emin olmak için veri çerçevesindeki Sample_ID’lere bakarız (örneğin, harflerin tümü büyük harf olması, harfler ve sayılar arasında fazladan alt çizgi olmaması, vb.)
head(sample_data$Sample_ID) # we again inspect only the first 6 using head()## [1] "S17BD05944" "S15BD07413" "S18BD07247" "S19BD07384" "S18BD07338" "S18BD02657"Ayrıca tüm örneklerin ağaç dosyasında bulunup bulunmadığını, eşleşip eşleşmedikleri bir DOĞRU veya YANLIŞ mantıksal vektörü oluşturarak karşılaştırabiliriz. (Basitleştirmek için burada gösterilmemiştir).
Bu vektörleri ağaçta olmayan herhangi bir örnek kimliğini göstermek için kullanabiliriz (hiçbiri yoktur).
sample_data$Sample_ID[!tree$tip.label %in% sample_data$Sample_ID]## character(0)İnceleme sonucu, veri çerçevesindeki Sample_ID formatının tip.labels’deki örnek isimlerine karşılık geldiğini görebiliriz. Bunların eşleşmesi için aynı sırada sıralanması gerekmez.
Devam etmeye hazırız!
38.3 Temel ağaç görselleştirmesi
Farklı ağaç düzenlemeleri
ggtree birçok farklı düzen formatı sunar ve bazıları özel amacınız için diğerlerinden daha uygun olabilir. Aşağıda birkaç gösterim var. Diğer seçenekler için bu çevrimiçi kitaba bakabilirsiniz.
İşte bazı örnek ağaç düzenleri:
ggtree(tree) # basit doğrusal ağaç
ggtree(tree, branch.length = "none") # uçların hizalandığı basit doğrusal ağaç
ggtree(tree, layout="circular") # basit dairesel ağaç
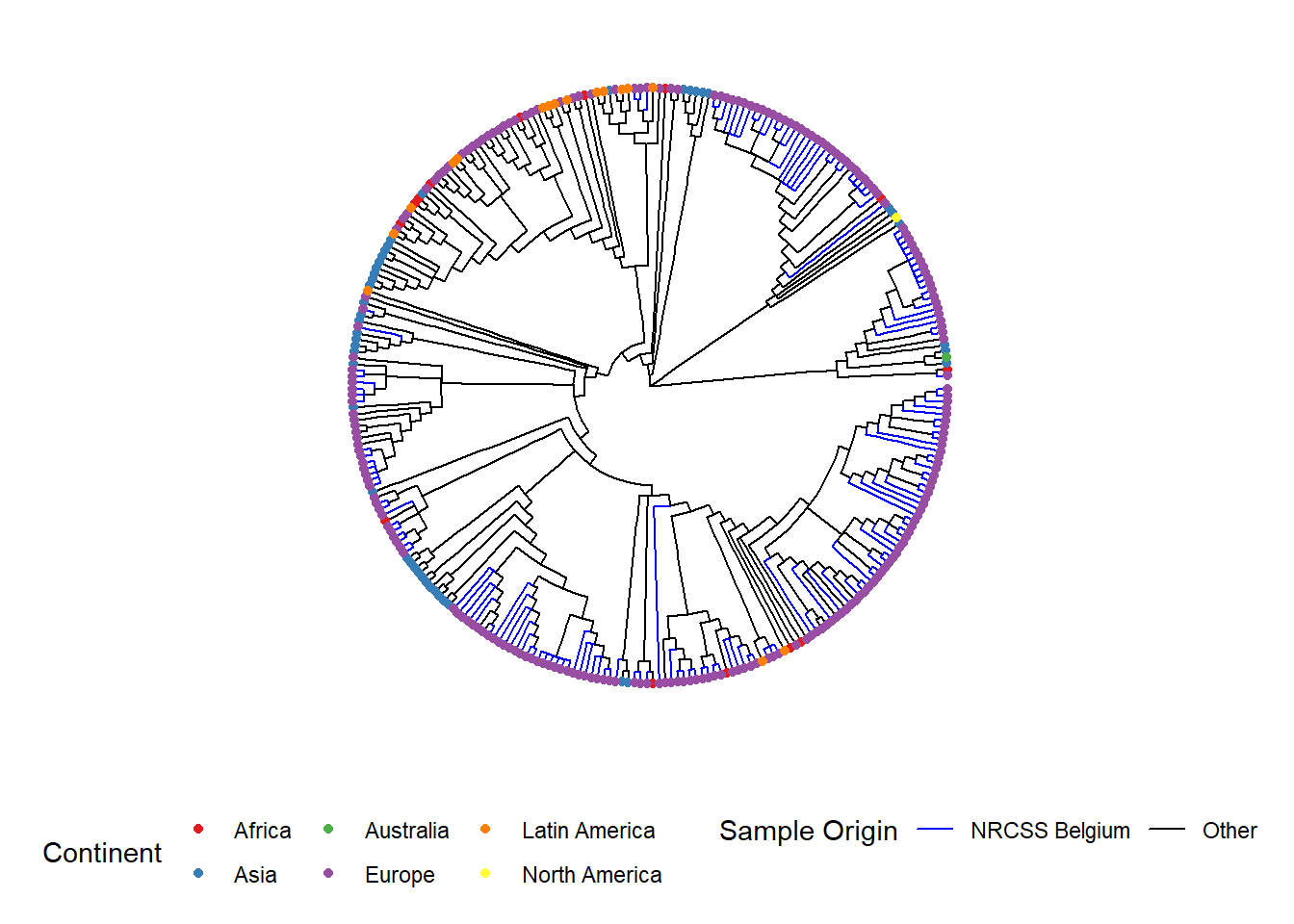
ggtree(tree, layout="circular", branch.length = "none") # uçların hizalandığı basit dairesel ağaç 


Örnek veriyi içeren basit ağaç
%<+% operatörü, sample_data veri çerçevesini ağaç dosyasına bağlamak için kullanılır. Ağacınızın en basit gösterimi, uçlara örnek isimlerin eklenmesi, ayrıca uç noktalarının ve istenirse dalların renklendirilmesidir:
İşte dairesel bir ağaç örneği:
ggtree(tree, layout = "circular", branch.length = 'none') %<+% sample_data + #%<+%, ağaca örnek verilerle veri çerçevesi ekler
aes(color = I(Belgium))+ # dalları veri çerçevenizdeki bir değişkene göre renklendirin
scale_color_manual(
name = "Sample Origin", # renk düzeninizin adı (lejantta bu şekilde görünecektir)
breaks = c("Yes", "No"), # değişkeninizdeki farklı seçenekler
labels = c("NRCSS Belgium", "Other"), # lejandınızda adı geçen farklı seçenekleri biçimlendirmenize olanak sağlar
values = c("blue", "black"), # değişkene atamak istediğiniz renk
na.value = "black") + # renkli NA değerleri siyahtır
new_scale_color()+ # başka bir değişken için ek bir renk şeması eklemeye izin verir
geom_tippoint(
mapping = aes(color = Continent), # kıtaya göre uç rengi. "shape =" ekleyerek şekli değiştirebilirsiniz.
size = 1.5)+ # uçtaki noktanın boyutunu tanımlayın
scale_color_brewer(
name = "Continent", # renk şemanızın adı (lejantta bu şekilde görünecektir)
palette = "Set1", # brewer paketiyle birlikte gelen bir dizi renk seçiyoruz
na.value = "grey") + # NA değerleri için gri rengi seçiyoruz
geom_tiplab2( # dalının ucuna numunenin adını ekler
color = 'black', # (+ ile istediğiniz kadar metin satırı ekleyin, ancak bunları yan yana yerleştirmek için ofset değerini ayarlamanız gerekebilir)
offset = 1,
size = 1,
geom = "text",
#align = TRUE
)+
ggtitle("Phylogenetic tree of Shigella sonnei")+ # grafiğinizin başlığı
theme(
axis.title.x = element_blank(), # x ekseni başlığını kaldırır
axis.title.y = element_blank(), # y ekseni başlığını kaldırır
legend.title = element_text( # açıklama başlığının yazı tipi boyutunu ve biçimini tanımlar
face = "bold",
size = 12),
legend.text=element_text( # gösterge metninin yazı tipi boyutunu ve biçimini tanımlar
face = "bold",
size = 10),
plot.title = element_text( # grafik başlığının yazı tipi boyutunu ve biçimini tanımlar
size = 12,
face = "bold"),
legend.position = "bottom", # lejant yerleşimini tanımlar
legend.box = "vertical", # lejant yerleşimini tanımlar
legend.margin = margin()) 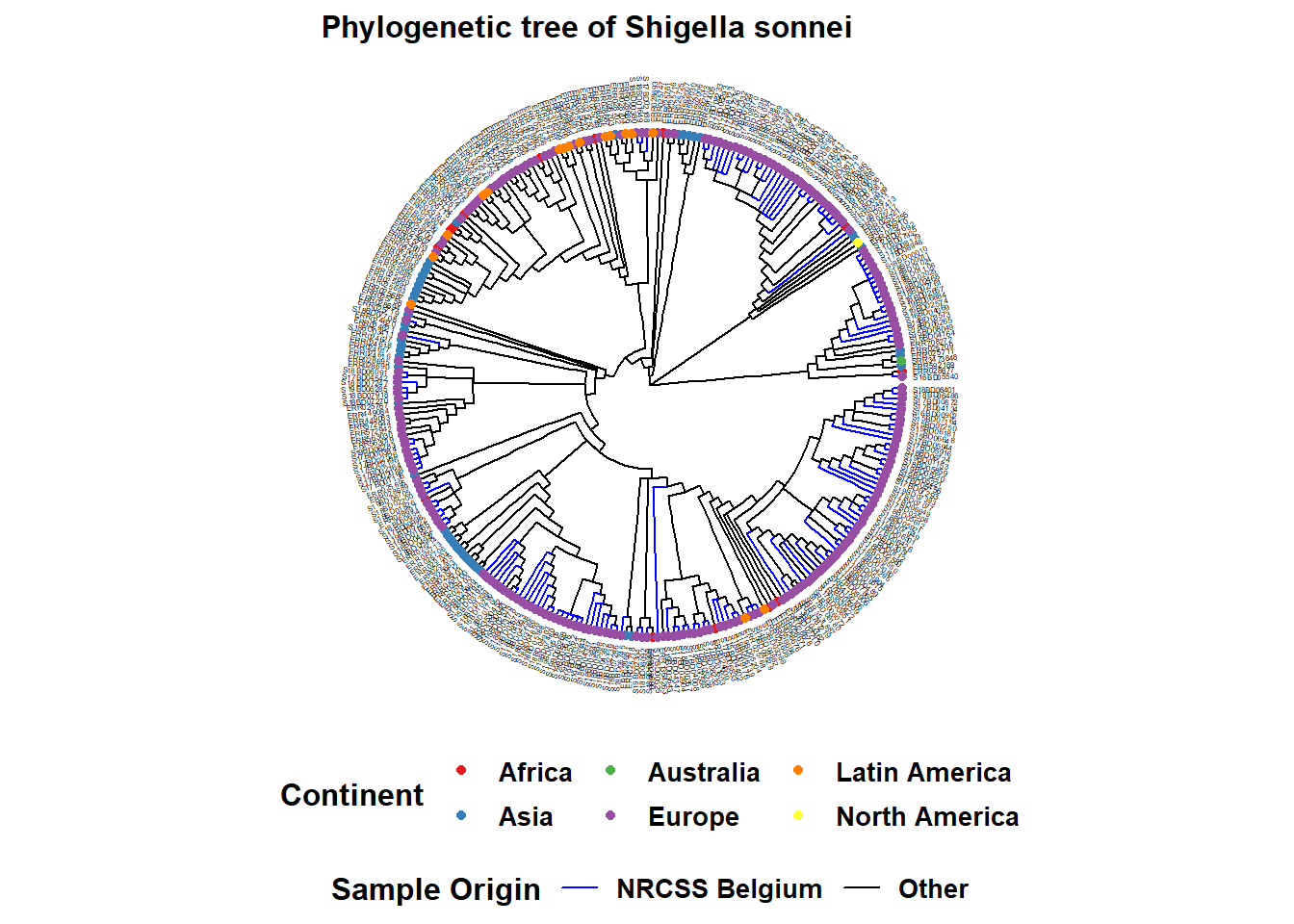
Diğer herhangi bir ggplot nesnesinde yaptığınız gibi, ağaç grafiğinizi ggsave() ile dışa aktarabilirsiniz. Bu şekilde yazıldığında, ggsave(), üretilen son grafiği belirttiğiniz dosya yoluna kaydeder. Alt klasörlere vb. kolayca kaydetmek için here() ve ilgili dosya yollarını kullanabileceğinizi unutmayın.
ggsave("example_tree_circular_1.png", width = 12, height = 14)38.4 Ağacın Manipülasyonu
Bazen çok büyük bir filogenetik ağacınız olabilir ve ağacın sadece bir kısmıyla ilgilenirsiniz. Örneğin, veri kümenizin büyük resimde nereye sığabileceğine dair geniş bir genel bakış elde etmek için tarihsel veya uluslararası örnekler içeren bir ağaç oluşturup daha sonra verilerinize daha yakından bakmak için daha büyük olan ağacın yalnızca o ilgilendiğiniz kısmını incelemek istersiniz.
Filogenetik ağaç dosyası yalnızca sekanslama veri analizinin çıktısı olduğundan, dosyanın kendisindeki nodların ve dalların sırasını değiştiremeyiz. Bunlar, ham NGS verilerinden önceki analizlerde zaten belirlenmiştir. Yine de parçaları yakınlaştırabiliyor, parçaları gizleyebiliyor ve hatta ağacın parçalarını alt kümelere ayırabiliyoruz.
Yakınlaştırmak
Ağacınızı “kesmek” istemiyorsanız ve sadece bir kısmını daha yakından incelemek istiyorsanız, belirli bir kısmı görüntülemek için yakınlaştırabilirsiniz.
İlk olarak, tüm ağacı doğrusal biçimde çiziyoruz ve ağaçtaki her bir noda sayısal etiketler ekliyoruz.
p <- ggtree(tree,) %<+% sample_data +
geom_tiplab(size = 1.5) + # ağaç dosyasındaki örnek adıyla tüm dalların uçlarını etiketler
geom_text2(
mapping = aes(subset = !isTip,
label = node),
size = 5,
color = "darkred",
hjust = 1,
vjust = 1) # ağaçtaki tüm nodları etiketler
p # çıktı alın
Belirli bir dalı yakınlaştırmak için (sağa doğru uzanarak), ggtree p nesnesinde viewClade() fonkisyonunu kullanın ve daha yakından bakmak için nod numarasını sağlayın:
viewClade(p, node = 452)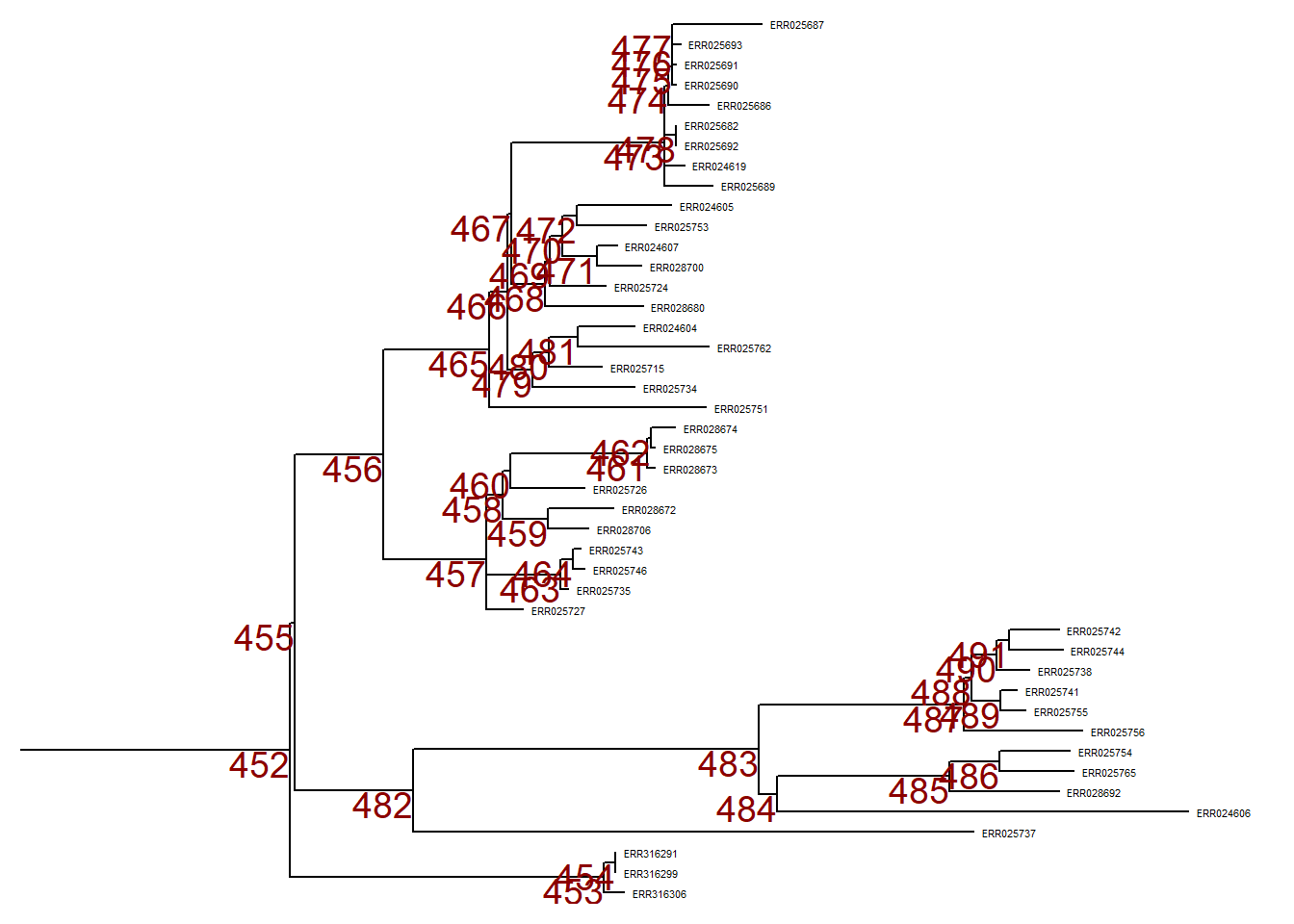
Daralan dallar
Ancak, bu dalı yok saymak isteyebiliriz ve aynı noddaki (nod nr. 452) collapse() fonksiyonunu kullanarak dalı daraltabiliriz. Bu ağaç p_collapsed olarak tanımlanır.
p_collapsed <- collapse(p, node = 452)
p_collapsed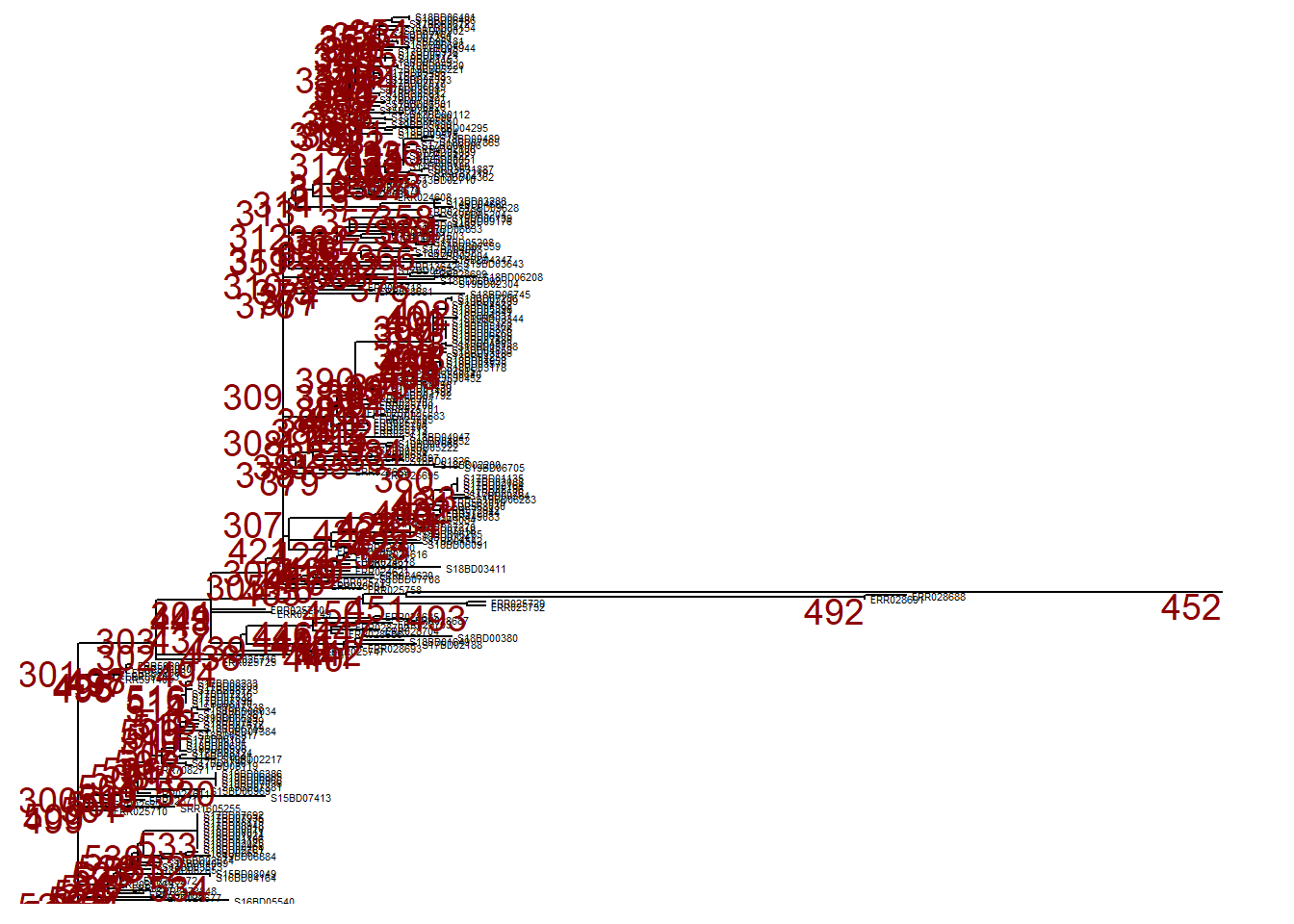
Anlaşılır olması için, p_collapsed’ı yazdırdığımızda, daraltılmış dalın noduna geom_point2() ile mavi elmas şekli ekleriz.
p_collapsed +
geom_point2(aes(subset = (node == 452)), # we assign a symbol to the collapsed node
size = 5, # define the size of the symbol
shape = 23, # define the shape of the symbol
fill = "steelblue") # define the color of the symbol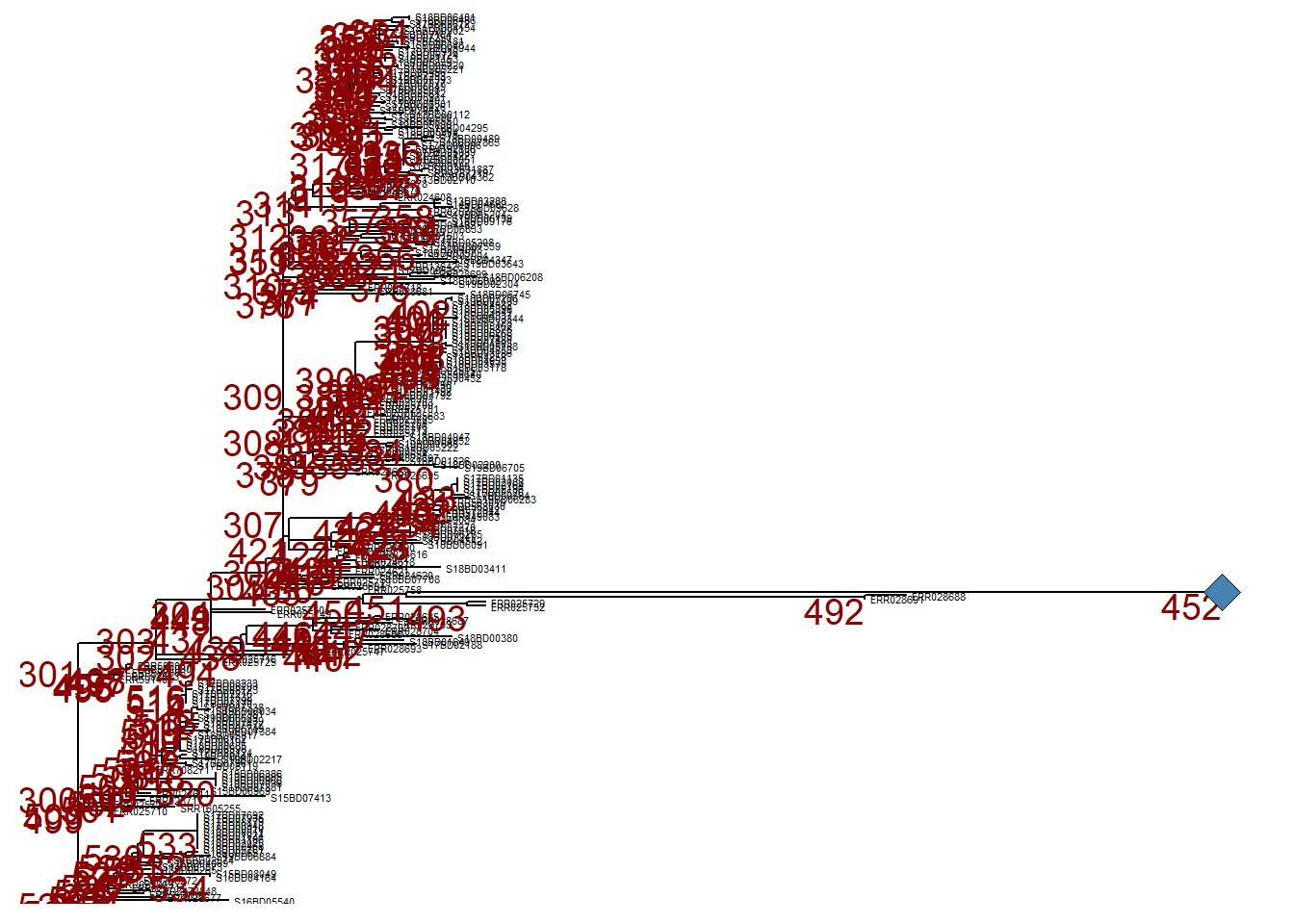
Ağacın alt kümelere ayrılması
Daha kalıcı bir değişiklik yapmak ya da çalışma için yeni ve küçültülmüş bir ağaç oluşturmak istiyorsak, ağacın bir kısmının tree_subset() ile alt kümesini alabiliriz. Ardından bu alt kümeyi yeni bir newick ağaç dosyası veya .txt dosyası olarak kaydedebilirsiniz.
İlk olarak, neyin altküme olacağına karar vermek için ağaç nodlarını ve uç etiketlerini inceleriz.
ggtree(
tree,
branch.length = 'none',
layout = 'circular') %<+% sample_data + # %<+% operatörünü kullanarak örnek verileri ekliyoruz
geom_tiplab(size = 1)+ # ağaç dosyasında örnek adıyla tüm dalların uçlarını etiketliyoruz
geom_text2(
mapping = aes(subset = !isTip, label = node),
size = 3,
color = "darkred") + # ağaçtaki tüm nodları etiketler
theme(
legend.position = "none", # lejantı uzaklaştırır
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(size = 12, face="bold"))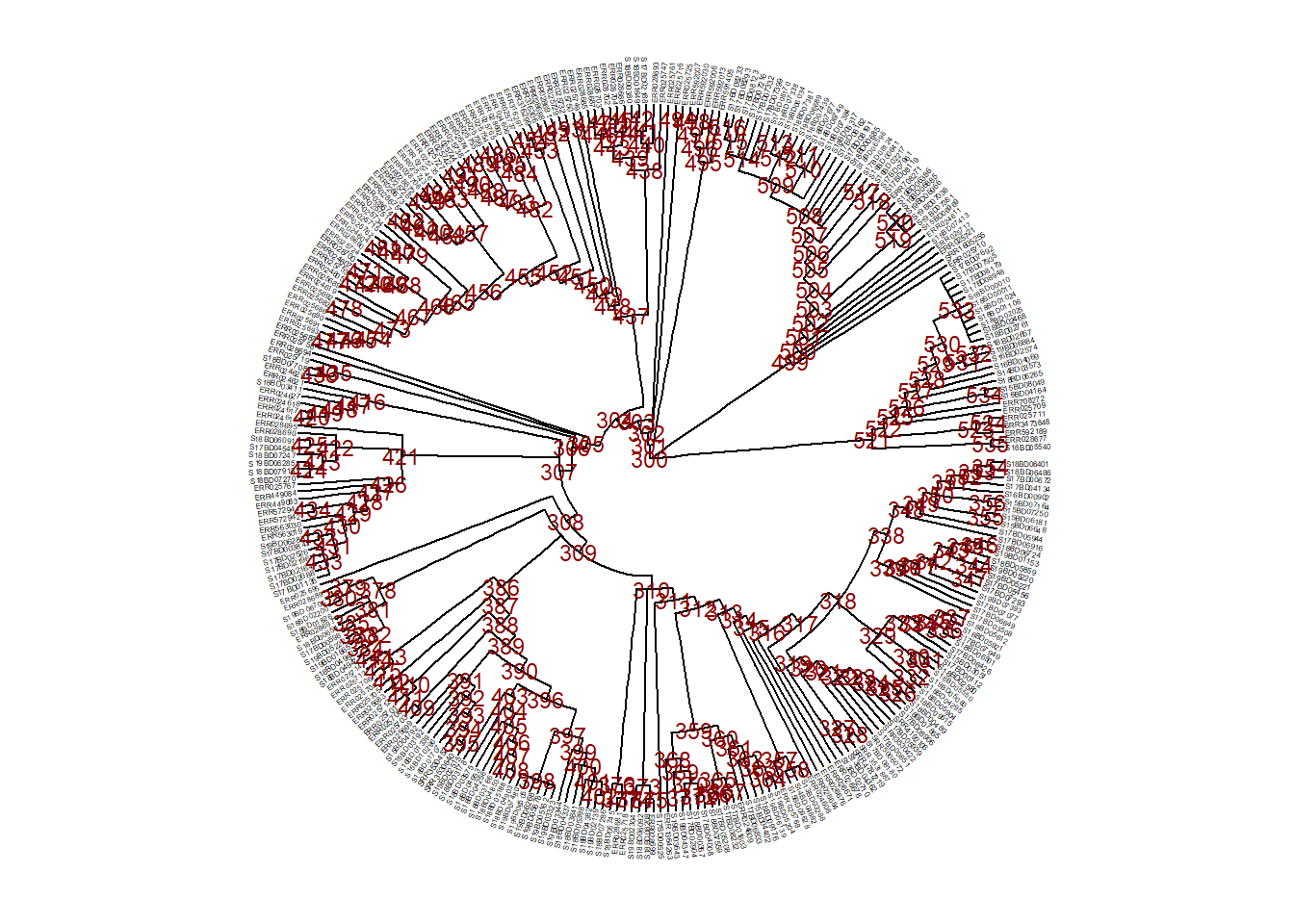
Şimdi, ağacın 528 numaralı nodundan alt küme oluşturmaya karar verdiğimizi varsayalım (sadece bu dalda 528 nodundan sonraki uçlarını tutalım) ve onu yeni bir sub_tree1 nesnesi olarak kaydedelim:
sub_tree1 <- tree_subset(
tree,
node = 528) # ağacın 528 nodundan alt kümesini oluşturuyoruz1 numaralı alt küme ağacına bir göz atalım:
ggtree(sub_tree1) +
geom_tiplab(size = 3) +
ggtitle("Subset tree 1")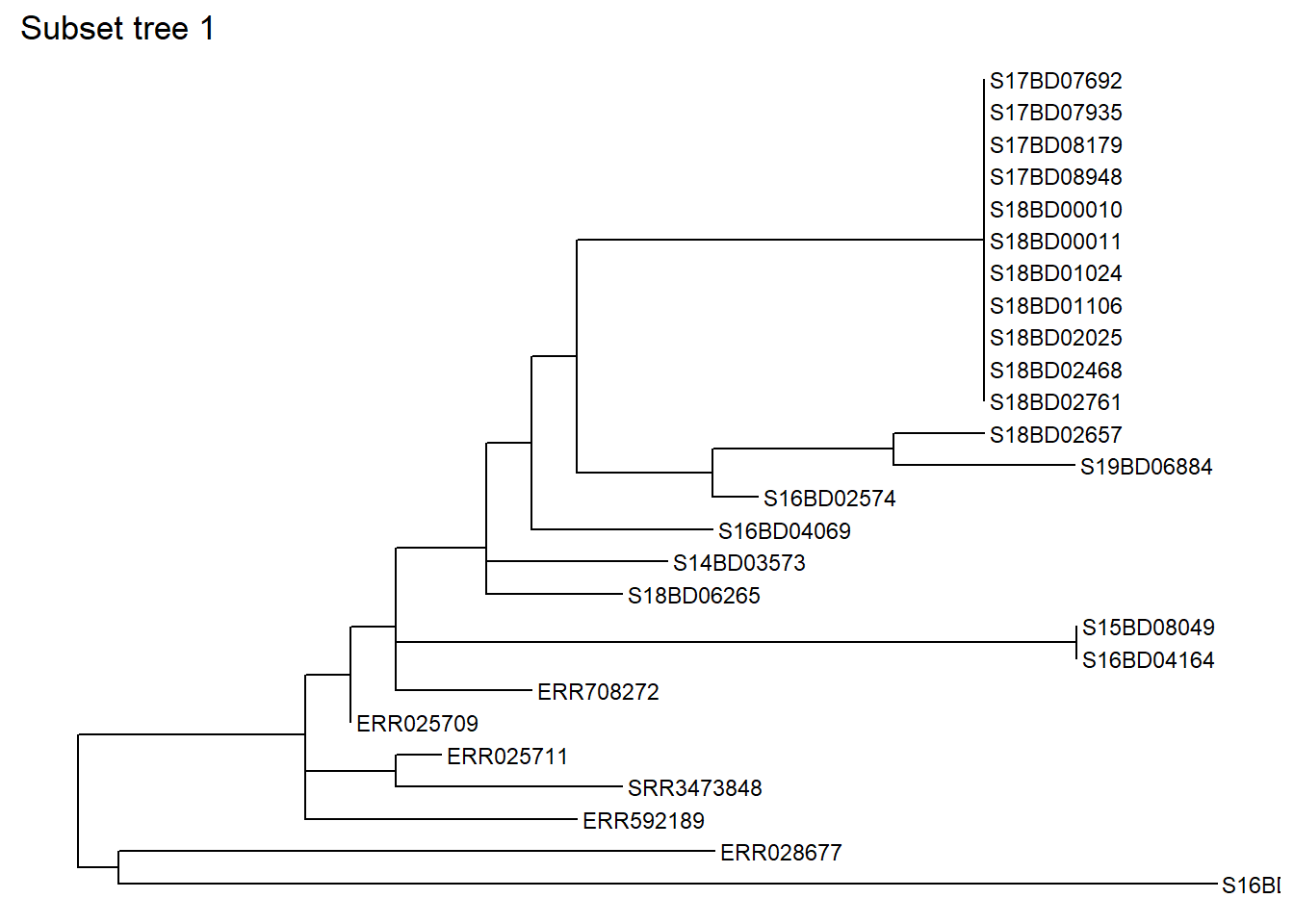
Ayrıca, “geriye” kaç tane nod eklemek istediğinizi belirterek belirli bir örneğe dayalı olarak alt kümeler oluşturabilirsiniz. Ağacın aynı kısmını bir örneğe dayanarak alt kümelendirelim, bu durumda S17BD07692 için 9 nod geriye giderek ve onu yeni bir sub_tree2 nesnesi olarak kaydedelim:
sub_tree2 <- tree_subset(
tree,
"S17BD07692",
levels_back = 9) # geri seviye sayısı, örnek ucundan geriye kaç nod gideceğinizi tanımlar.Şimdi alt küme ağacına bir göz atalım:
ggtree(sub_tree2) +
geom_tiplab(size =3) +
ggtitle("Subset tree 2")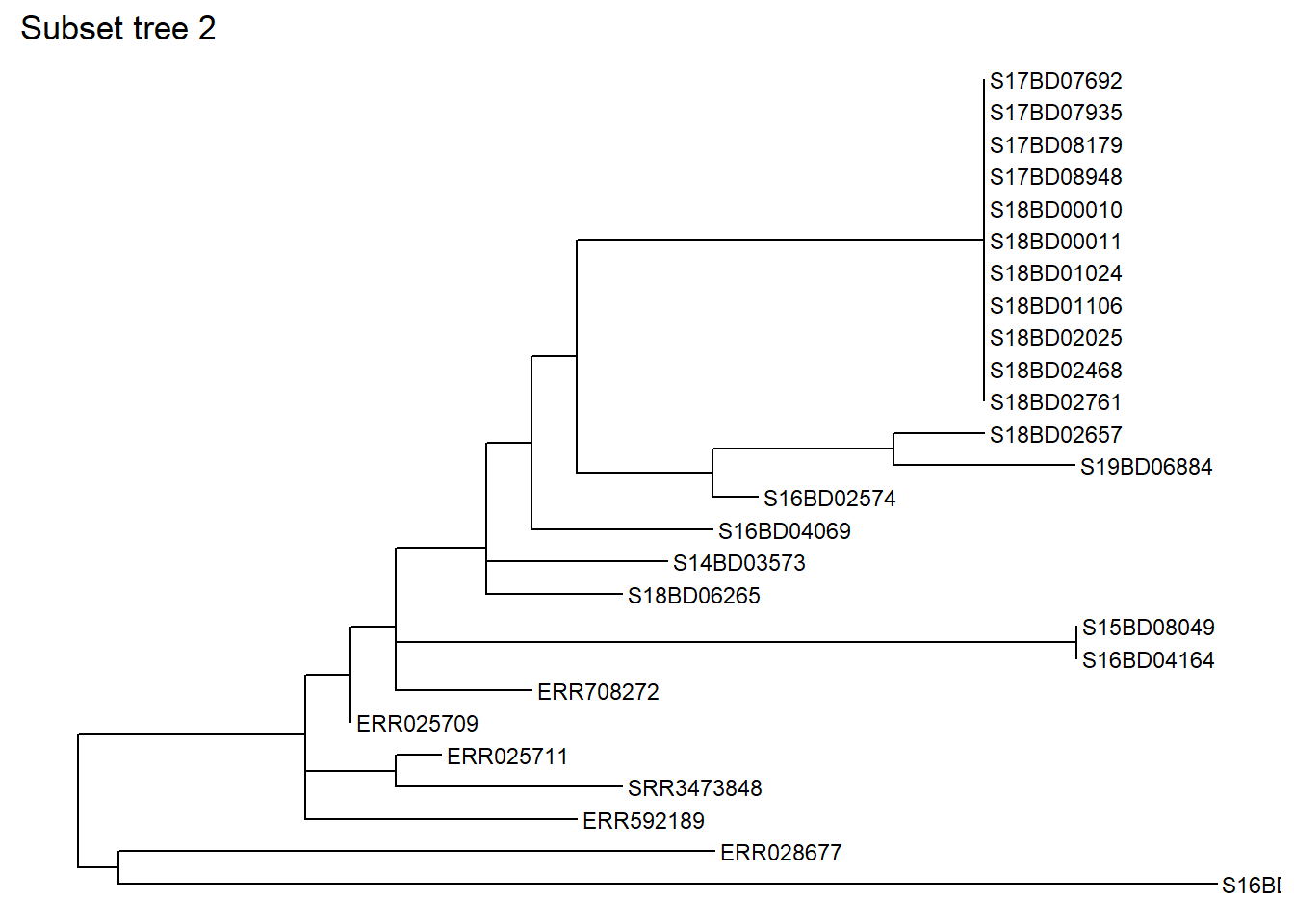
Yeni ağacınızı, ape paketindeki write.tree() fonksiyonunu kullanarak bir Newick formatında veya hatta bir metin dosyası olarak da kaydedebilirsiniz
# .nwk formatında kaydetmek için
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.nwk')
# .txt formatında kaydetmek için
ape::write.tree(sub_tree2, file='data/phylo/Shigella_subtree_2.txt')Ağaçtaki nodları döndürmek
Daha önce de belirtildiği gibi, ağaçtaki uçların veya nodların sırasını değiştiremeyiz, çünkü bu onların genetik akrabalıklarına dayanır ve görsel manipülasyona izin yoktur. Ancak, görselleştirmemizi kolaylaştıracaksa, dalları nodların etrafında döndürebiliriz.
İlk olarak, işlemek istediğimiz nodu seçmek için yeni alt küme ağacımızı (alt küme 2) nod etiketleriyle çizeriz ve bir ggtree çizim nesnesi ‘p’ olarak saklarız.
p <- ggtree(sub_tree2) +
geom_tiplab(size = 4) +
geom_text2(aes(subset=!isTip, label=node), # ağaçtaki tüm nodları etiketler
size = 5,
color = "darkred",
hjust = 1,
vjust = 1)
p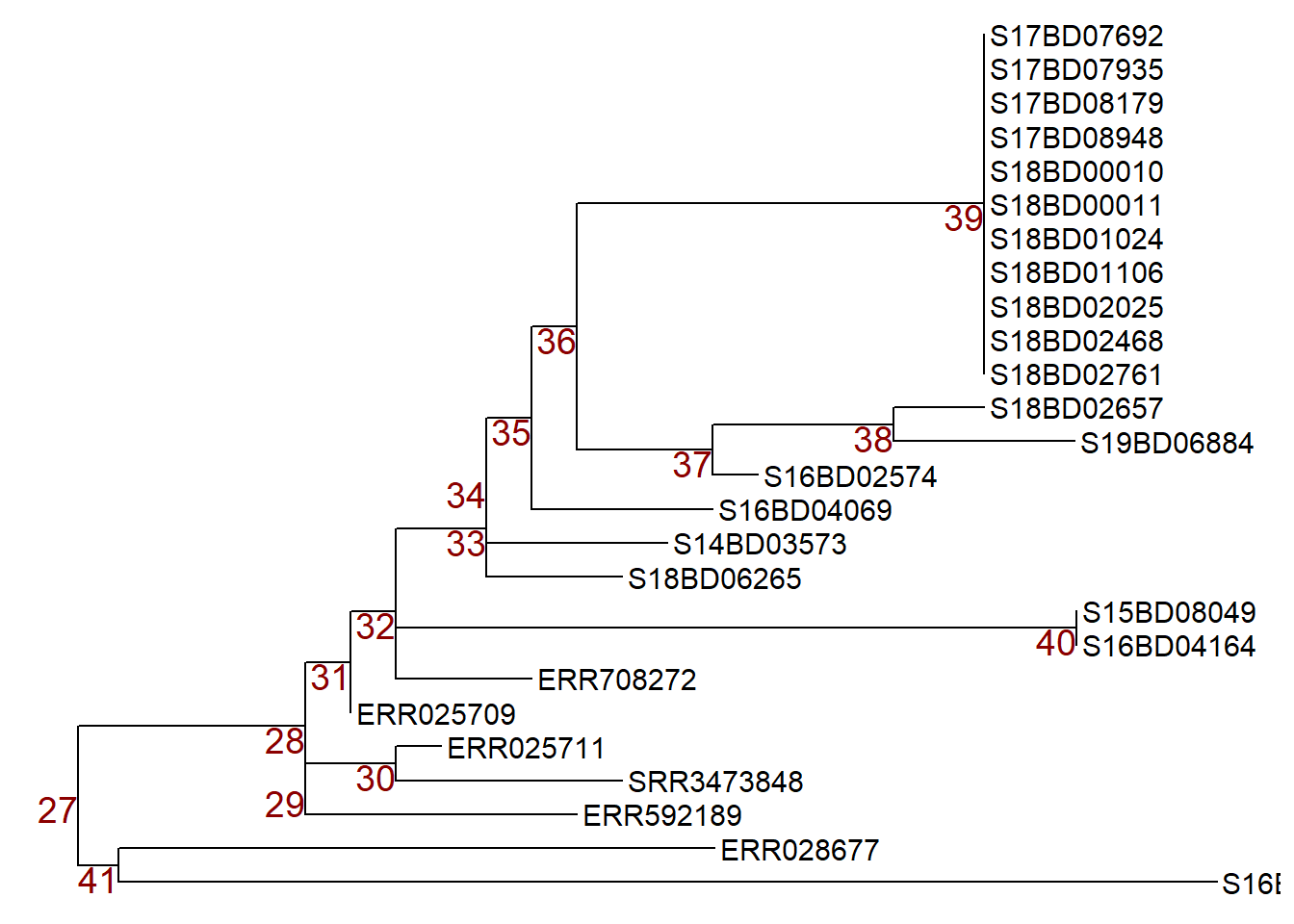
Ardından, ggtree::rotate() veya ggtree::flip() fonksiyonlarını uygulayarak nodları değiştirebiliriz: Not: Hangi nodları manuple ettiğimizi göstermek için, ilgilendiğimiz noddaki örnekleri vurgulamak için önce ggtree’den geom_hilight() fonksiyonunu uygularız. ggtree çizim nesnesini yeni bir nesne olarak (p1) saklayın.
p1 <- p + geom_hilight( # nod 39'u mavi olarak vurgular, "extend =" renk bloğunun uzunluğunu tanımlamamızı sağlar
node = 39,
fill = "steelblue",
extend = 0.0017) +
geom_hilight( # 37 numaralı düğümü sarı renkle vurgular
node = 37,
fill = "yellow",
extend = 0.0017) +
ggtitle("Original tree")
p1 # çıktı alın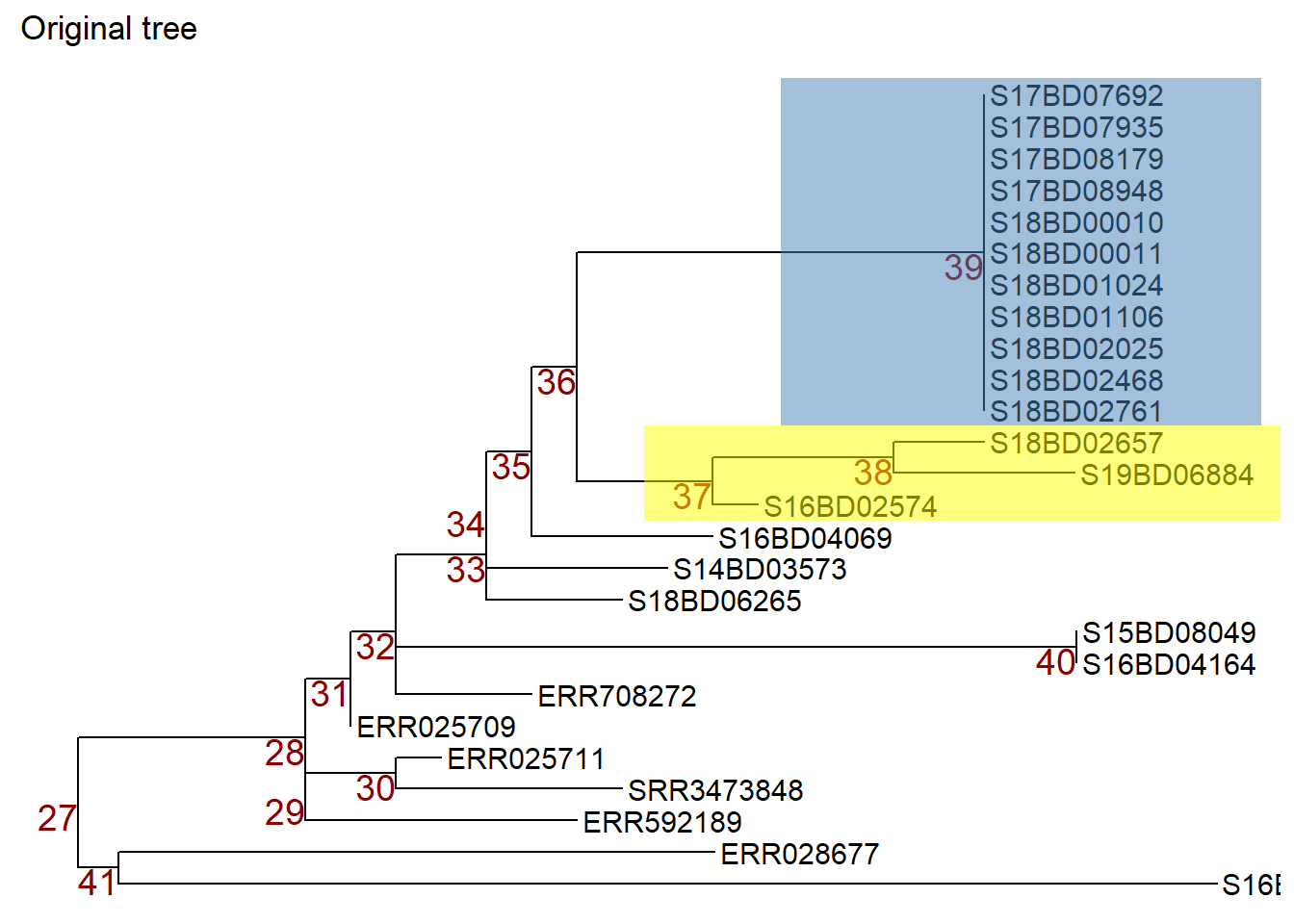
Şimdi p1 nesnesindeki nod 37’yi döndürebiliriz, böylece nod 38’deki örnekler en üste hareket eder. Döndürülen ağacı yeni bir p2 nesnesi olarak saklıyoruz.
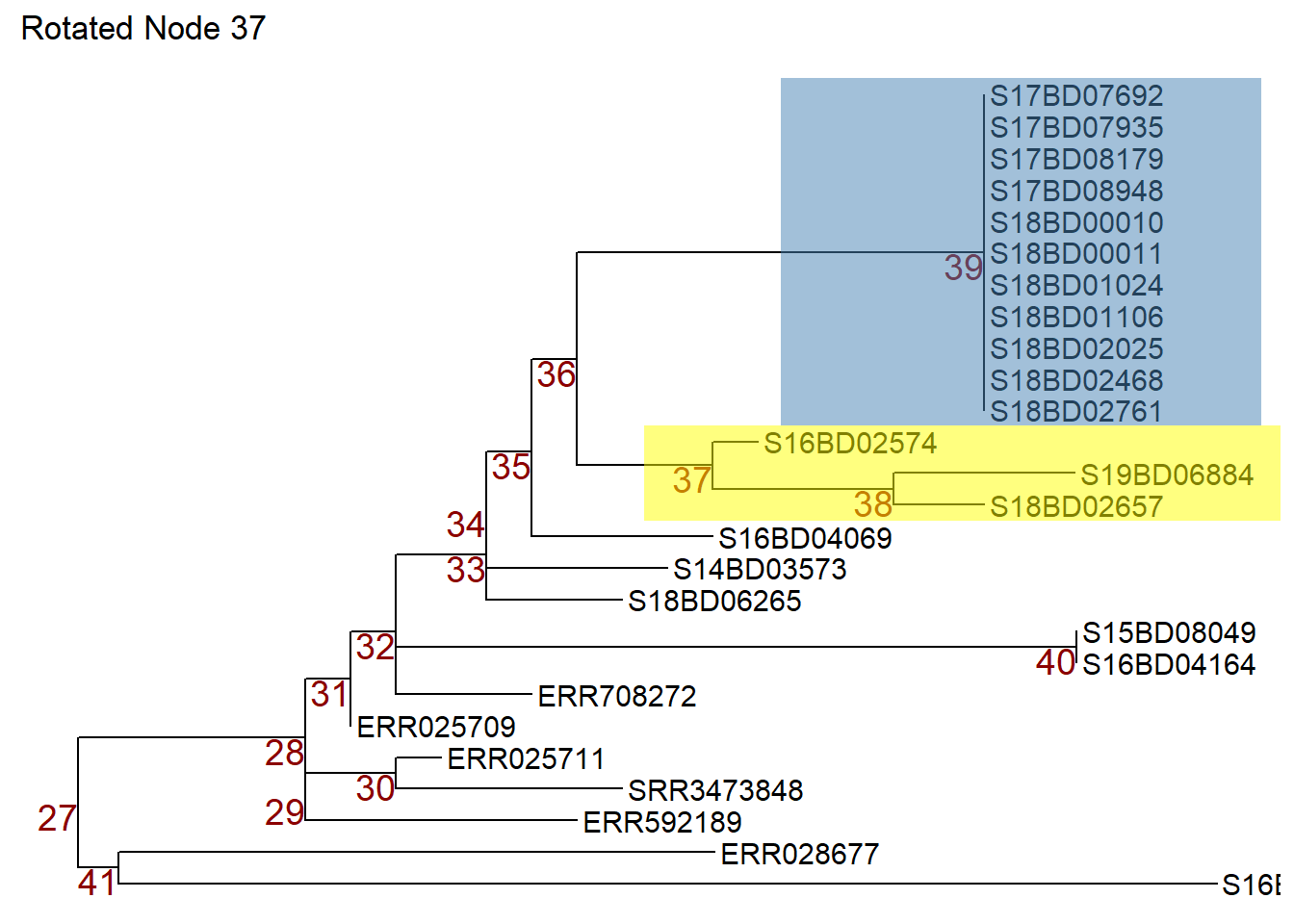
Ya da p1 nesnesindeki 36 nolu nodu döndürmek ve 37 nolu nodu yukarı ve 39 nolu nodu aşağıya çevirmek için flip (çevirme= komutunu kullanabiliriz. Ters çevrilmiş ağacı yeni bir p3 nesnesi olarak saklıyoruz.
p3 <- flip(p1, 39, 37) +
ggtitle("Rotated Node 36")
p3 # çıktı alın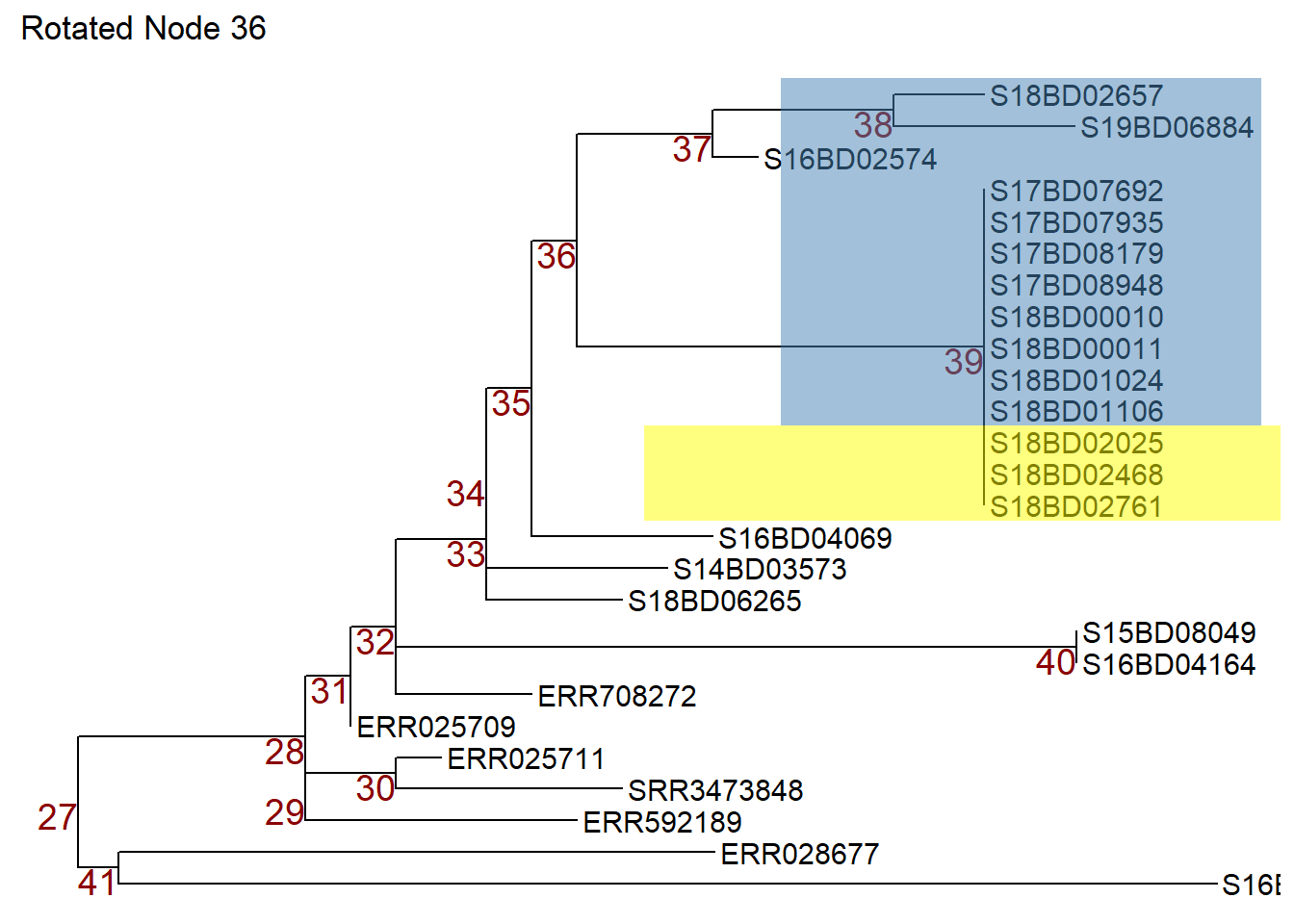
Numune verilerini içeren açıklamalı örnek alt ağaç
2017 ve 2018’de alt ağacımızda 39. nodda meydana gelen klonal genişlemeli vaka kümesini araştırdığımızı varsayalım. Yakın ilişkili diğer suşların kökenini görmek için suş izolasyonunun yanı sıra ülkeye göre seyahat geçmişi ve rengi de ekliyoruz:
ggtree(sub_tree2) %<+% sample_data + # sample_data'ya bağlanmak için %<+% operatörünü kullanırız
geom_tiplab( # ağaç dosyasındaki numune adıyla tüm dalların uçlarını etiketler
size = 2.5,
offset = 0.001,
#align = TRUE
) +
theme_tree2()+
xlim(0, 0.015)+ # ağacımızın x ekseni sınırlarını ayarlar
geom_tippoint(aes(color=Country), # uç noktasını kıtaya göre renklendir
size = 1.5)+
scale_color_brewer(
name = "Country",
palette = "Set1",
na.value = "grey")+
geom_tiplab( # uçlara bir metin etiketi olarak izolasyon yılı ekleyin
aes(label = Year),
color = 'blue',
offset = 0.0045,
size = 3,
linetype = "blank" ,
geom = "text",
#align = TRUE
)+
geom_tiplab( # uçlara kırmızı renkte bir metin etiketi olarak seyahat geçmişi ekleyin
aes(label = Travel_history),
color = 'red',
offset = 0.006,
size = 3,
linetype = "blank",
geom = "text",
#align = TRUE
)+
ggtitle("Phylogenetic tree of Belgian S. sonnei strains with travel history")+ # grafik başlığı ekle
xlab("genetic distance (0.001 = 4 nucleotides difference)")+ # x eksenine bir etiket ekleyin
theme(
axis.title.x = element_text(size = 10),
axis.title.y = element_blank(),
legend.title = element_text(face = "bold", size = 12),
legend.text = element_text(face = "bold", size = 10),
plot.title = element_text(size = 12, face = "bold"))
Gözlemimiz, yıllar içinde Belçika’da dolaşan ve en son salgınımıza neden olan Asya’dan kaynaklanan suşa işaret ediyor.
Daha karmaşık ağaçlar: Örnek verilerin ısı haritalarını ekleme
ggtree::gheatmap() fonksiyonunu kullanarak bir ısı haritası biçiminde antimikrobiyal direnç genlerinin kategorik olarak varlığı ve antimikrobiyallere karşı ölçülen direnç için sayısal değerler gibi daha karmaşık bilgiler de ekleyebiliriz.
İlk önce ağacımızı çizmemiz gerekiyor (bu doğrusal veya dairesel olabilir) ve ağacı yeni bir ggtree çizim nesnesi p’de saklamamız gerekiyor: Bölüm 3’teki sub_tree’yi kullanacağız.)
p <- ggtree(sub_tree2, branch.length='none', layout='circular') %<+% sample_data +
geom_tiplab(size =3) +
theme(
legend.position = "none",
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(
size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p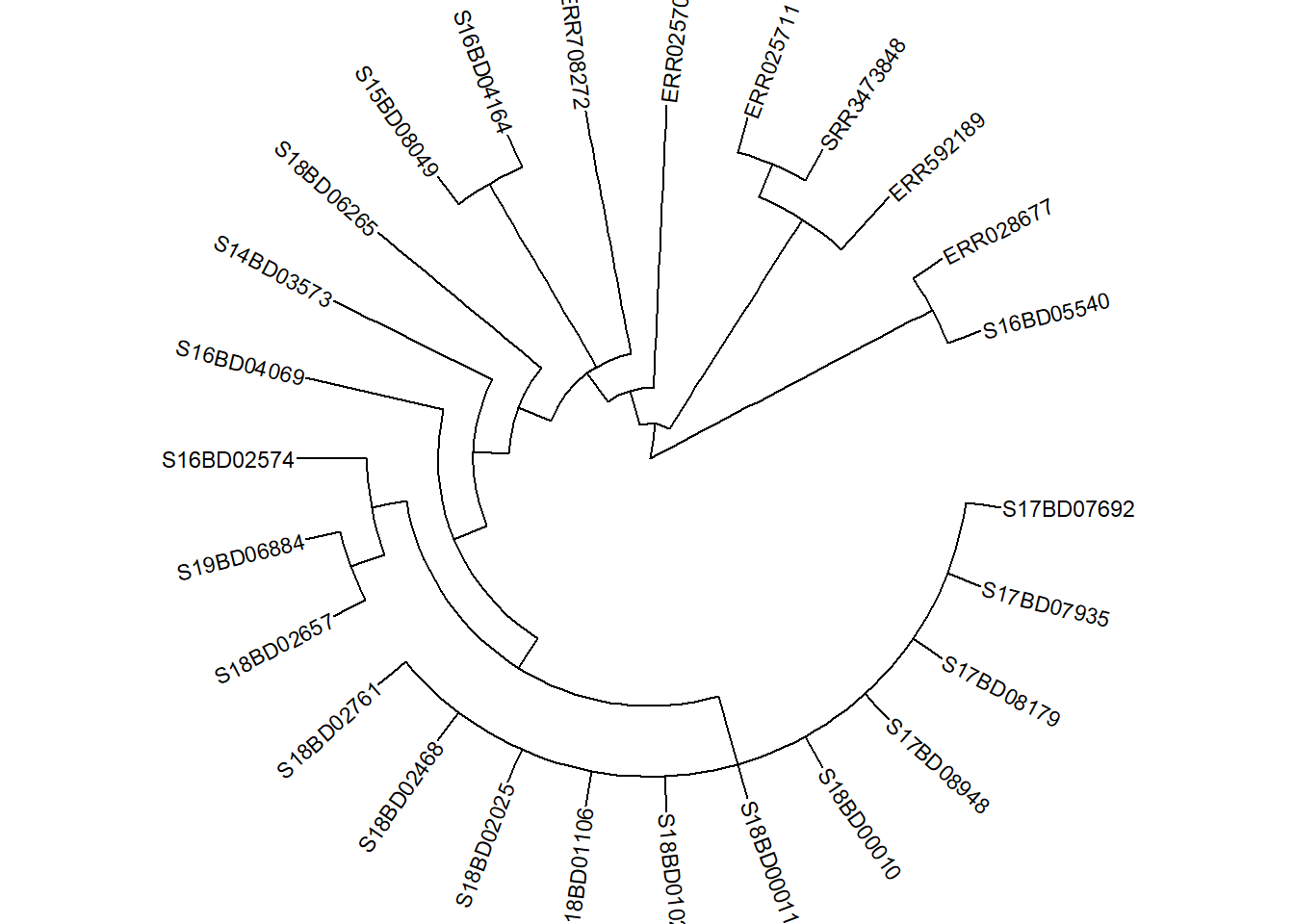
İkinci olarak verilerimizi hazırlıyoruz. Farklı değişkenleri yeni renk şemaları ile görselleştirmek için veri çerçevemizi istenen değişkene göre alt gruplara ayırıyoruz. Sample_ID’yi satır adları olarak eklemek önemlidir, aksi takdirde verileri ağaç tip.labels ile eşleştiremez:
Örneğimizde Shigella enfeksiyonlarını tedavi etmek için kullanılan önemli bir birinci basamak antibiyotik olan siprofloksasine direnç kazandırabilecek mutasyonlara ve cinsiyete bakmak istiyoruz.
Cinsiyet için bir veri çerçevesi oluşturuyoruz:
gender <- data.frame("gender" = sample_data[,c("Gender")])
rownames(gender) <- sample_data$Sample_IDSiprofloksasin direncine nede olan gyrA genindeki mutasyonlar için bir veri çerçevesi oluşturuyoruz:
cipR <- data.frame("cipR" = sample_data[,c("gyrA_mutations")])
rownames(cipR) <- sample_data$Sample_IDSiprofloksasin için ölçülen minimum inhibitör konsantrasyonu (MIC) için bir veri çerçevesi oluşturuyoruz:
MIC_Cip <- data.frame("mic_cip" = sample_data[,c("MIC_CIP")])
rownames(MIC_Cip) <- sample_data$Sample_IDFilogenetik ağaca cinsiyet için ikili bir ısı haritası ekleyen ve onu yeni bir ggtree grafi nesnesi h1’de saklayan bir ilk grafik oluşturuyoruz:
h1 <- gheatmap(p, gender, # ağaç grafiğimize cinsiyet veri çerçevesinin ısı haritası katmanını ekliyoruz
offset = 10, # ofset, ısı haritasını sağa kaydırır,
width = 0.10, # genişlik, ısı haritası sütununun genişliğini tanımlar,
color = NULL, # renk, ısı haritası sütunlarının kenarlığını tanımlar
colnames = FALSE) + # ısı haritası için sütun adlarını gizler
scale_fill_manual(name = "Gender", # cinsiyet için renklendirme şemasını ve lejandı tanımlayın
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h1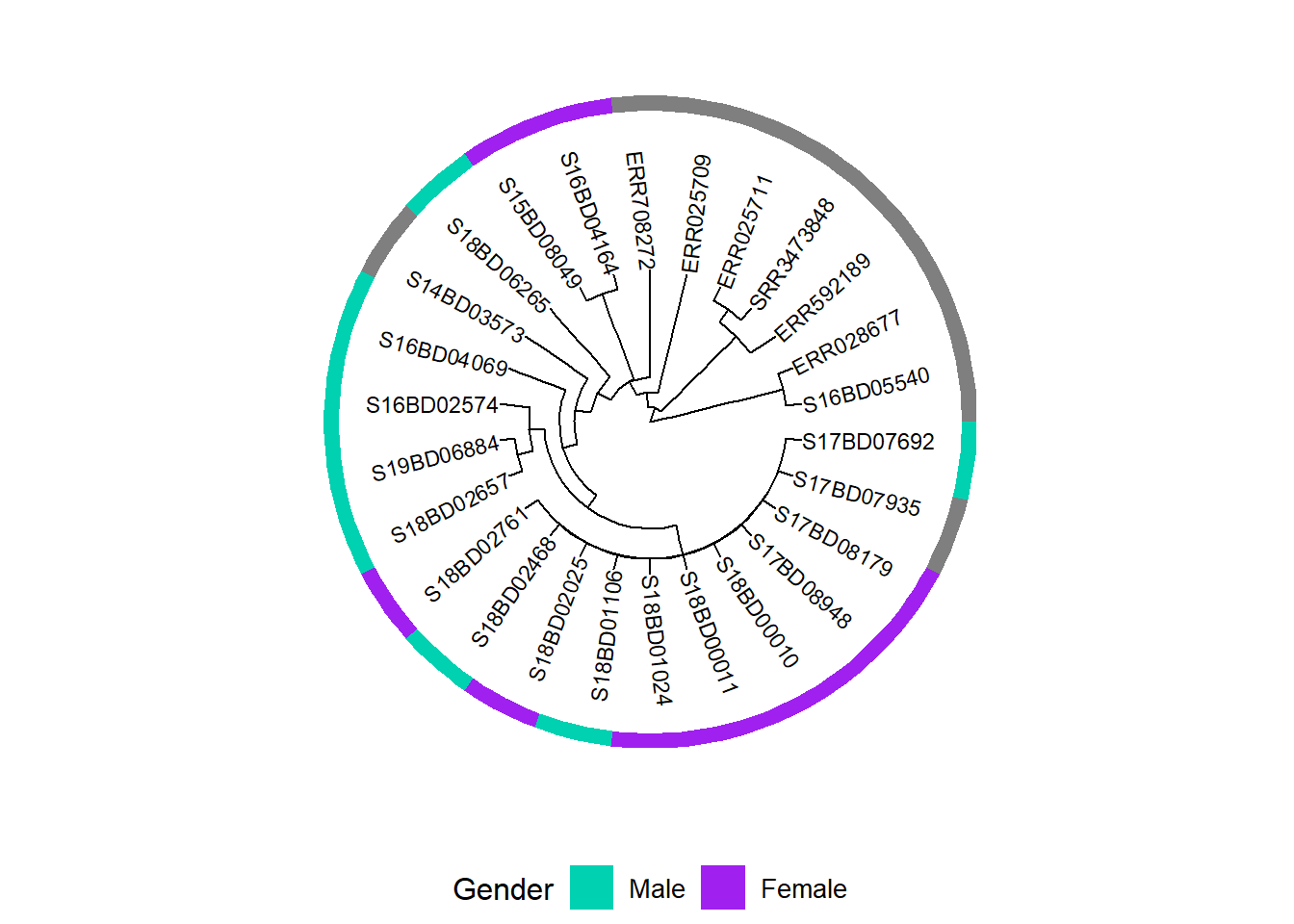
Ardından, gyrA genindeki siprofloksasine direnç sağlayan mutasyonlar hakkında bilgi ekliyoruz:
Not: WGS verilerinde kromozomal nokta mutasyonlarının varlığı, Zankari ve arkadaşları tarafından geliştirilen PointFinder aracı kullanılarak önceden belirlenmiştir. (referanslar bölümündeki referansa bakın)
İlk olarak, mevcut çizim nesnemiz h1’e yeni bir renk şeması atarız ve onu nesne h2’de saklarız. Bu, ısı haritasındaki ikinci değişkenimizin renklerini tanımlamamızı ve değiştirmemizi sağlar.
h2 <- h1 + new_scale_fill() Ardından ikinci ısı haritası katmanını h2’ye ekler ve birleştirilmiş grafikleri yeni bir nesne h3’te saklarız:
h3 <- gheatmap(h2, cipR, # Siprofloksasine direnç mutasyonlarını tanımlayan ikinci ısı haritası satırını ekler
offset = 12,
width = 0.10,
colnames = FALSE) +
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l")) +
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h3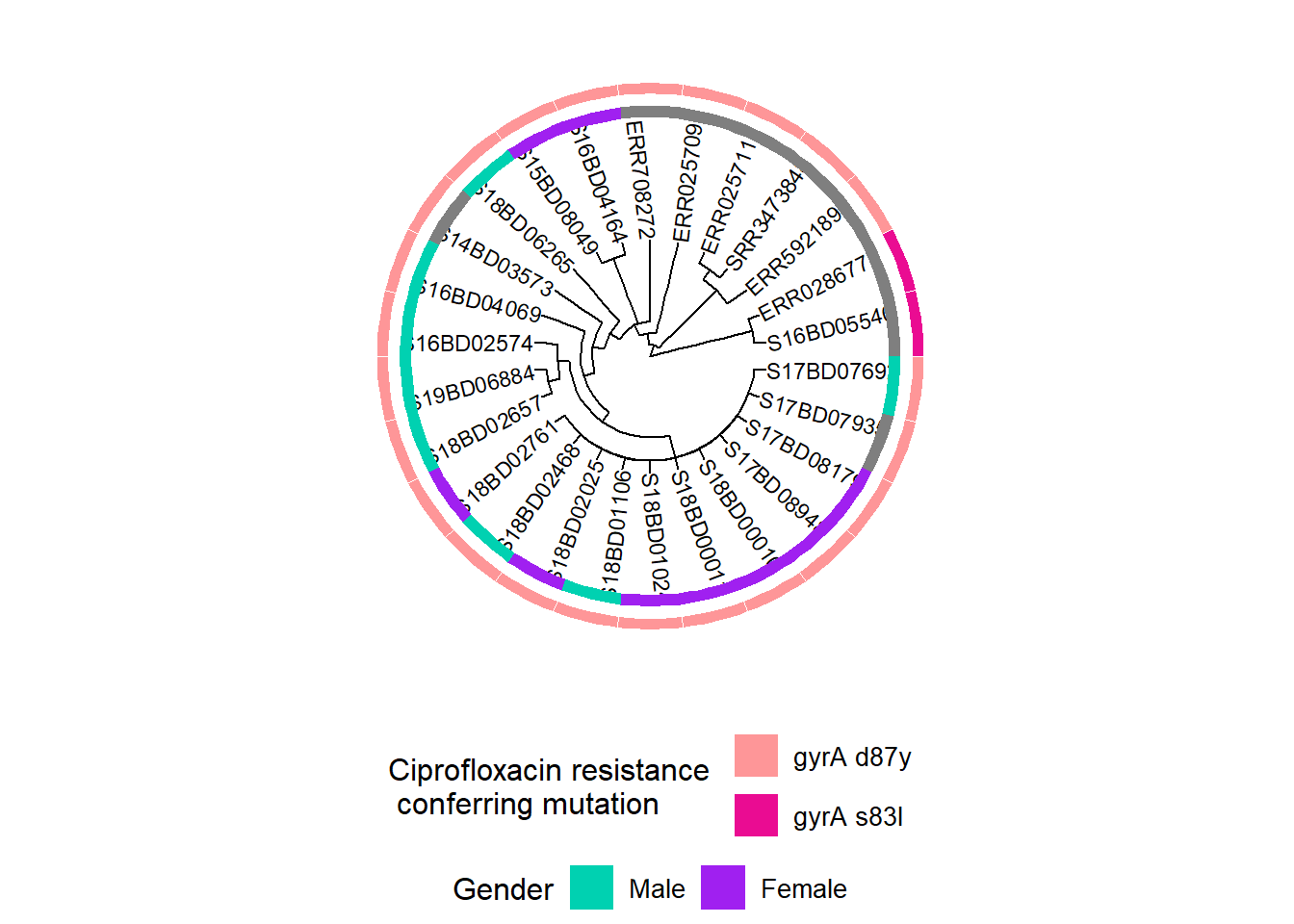
İlk önce mevcut nesne h3’e yeni bir renk ölçeği katmanı ekleyerek ve ardından nihai nesne h5’i üretmek için elde edilen h4 nesnesine her suş için siprofloksasin için minimum inhibitör konsantrasyonu (MIC) verilerini ekleyerek yukarıdaki işlemi tekrarlıyoruz. :
# İlk önce yeni renklendirme şemasını ekliyoruz:
h4 <- h3 + new_scale_fill()
# sonra ikisini yeni bir grafikte birleştiriyoruz:
h5 <- gheatmap(h4, MIC_Cip,
offset = 14,
width = 0.10,
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin", # burada MIC'in sürekli değişkeni için bir gradyan renk şeması tanımlıyoruz
low = "yellow", high = "red",
breaks = c(0, 0.50, 1.00),
na.value = "white") +
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h5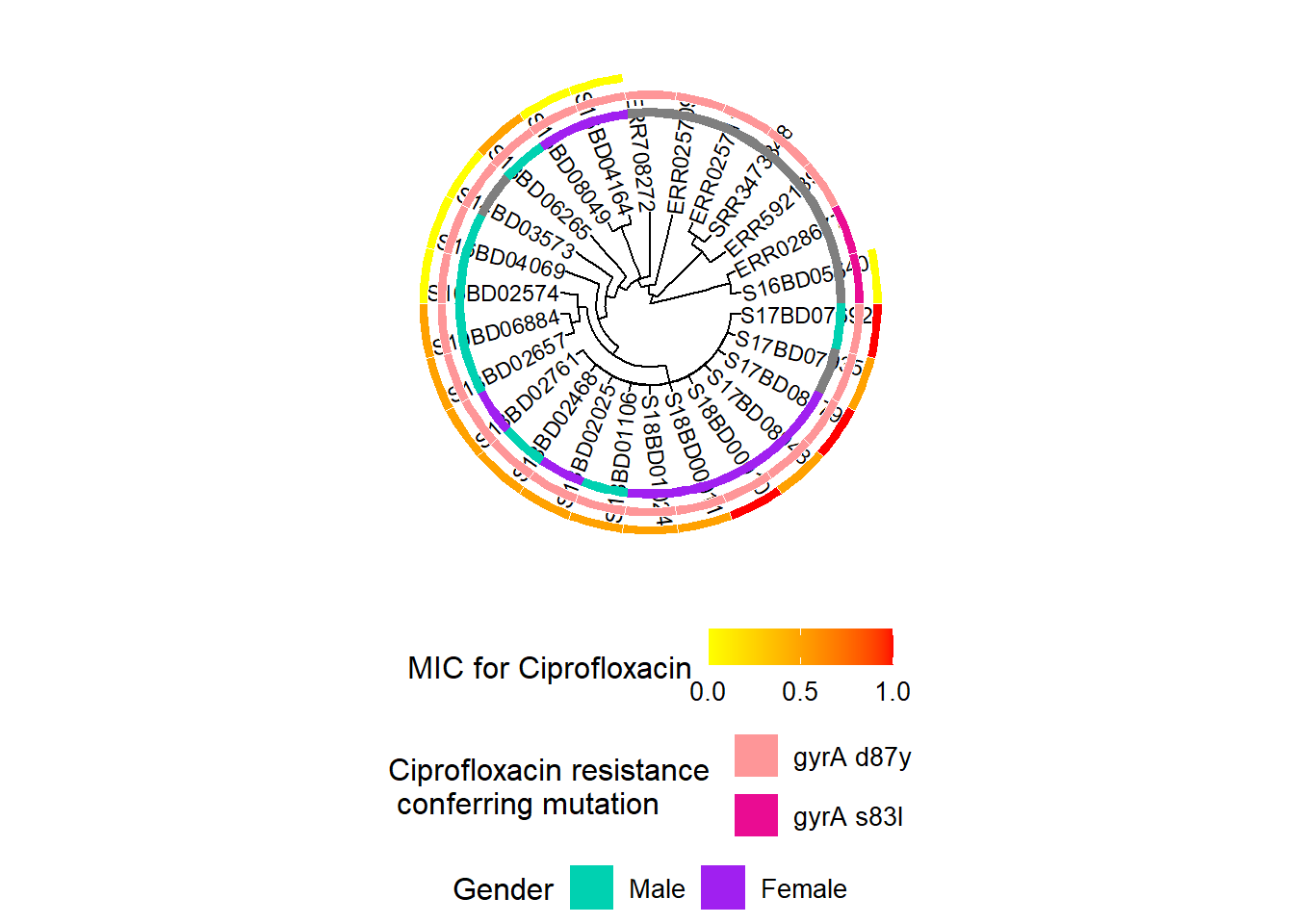
Aynı alıştırmayı doğrusal bir ağaç için de yapabiliriz:
p <- ggtree(sub_tree2) %<+% sample_data +
geom_tiplab(size = 3) + # labels the tips
theme_tree2()+
xlab("genetic distance (0.001 = 4 nucleotides difference)")+
xlim(0, 0.015)+
theme(legend.position = "none",
axis.title.y = element_blank(),
plot.title = element_text(size = 12,
face = "bold",
hjust = 0.5,
vjust = -15))
p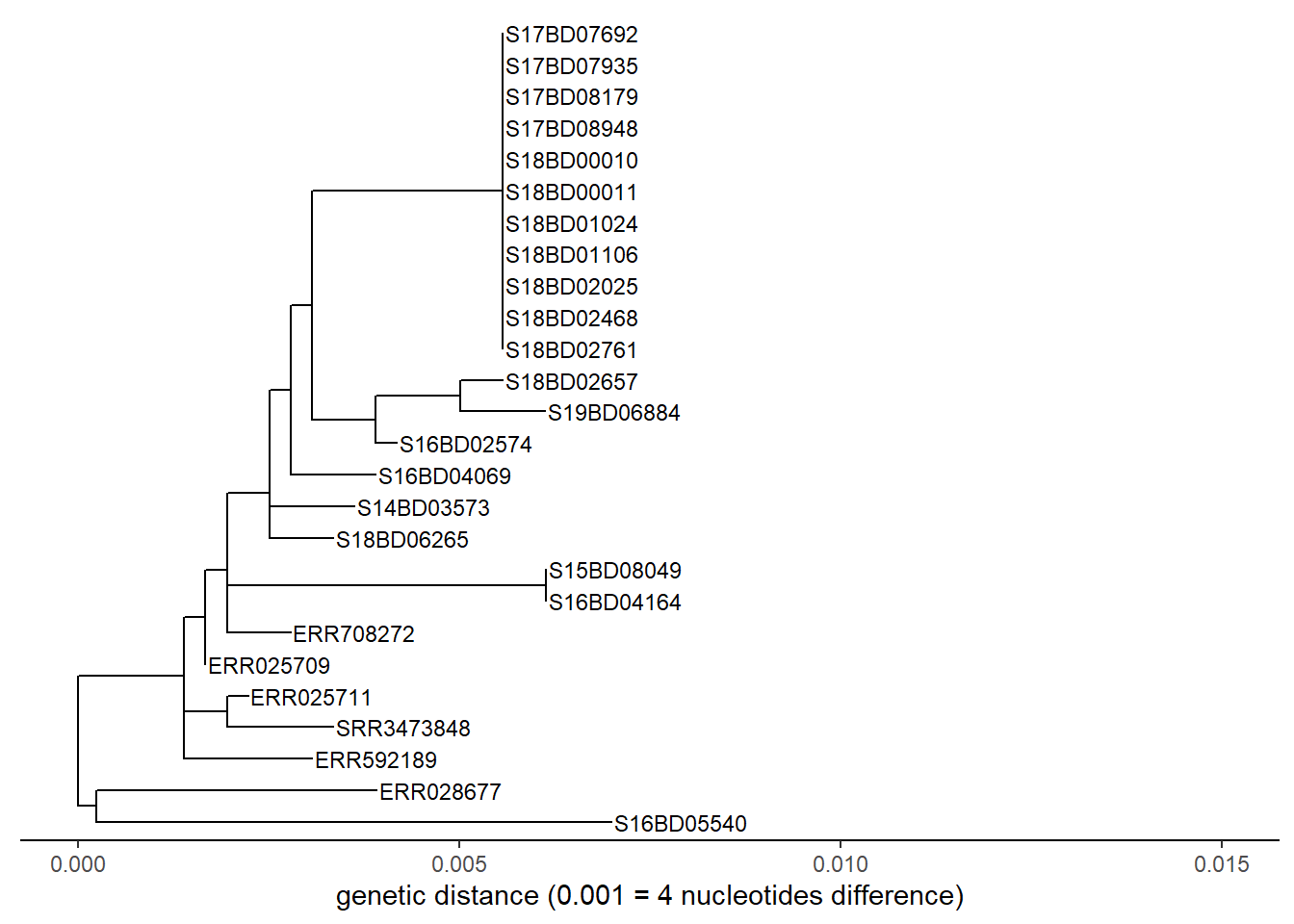
Öncelikle cinsiyeti giriyoruz
h1 <- gheatmap(p, gender,
offset = 0.003,
width = 0.1,
color="black",
colnames = FALSE)+
scale_fill_manual(name = "Gender",
values = c("#00d1b1", "purple"),
breaks = c("Male", "Female"),
labels = c("Male", "Female"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h1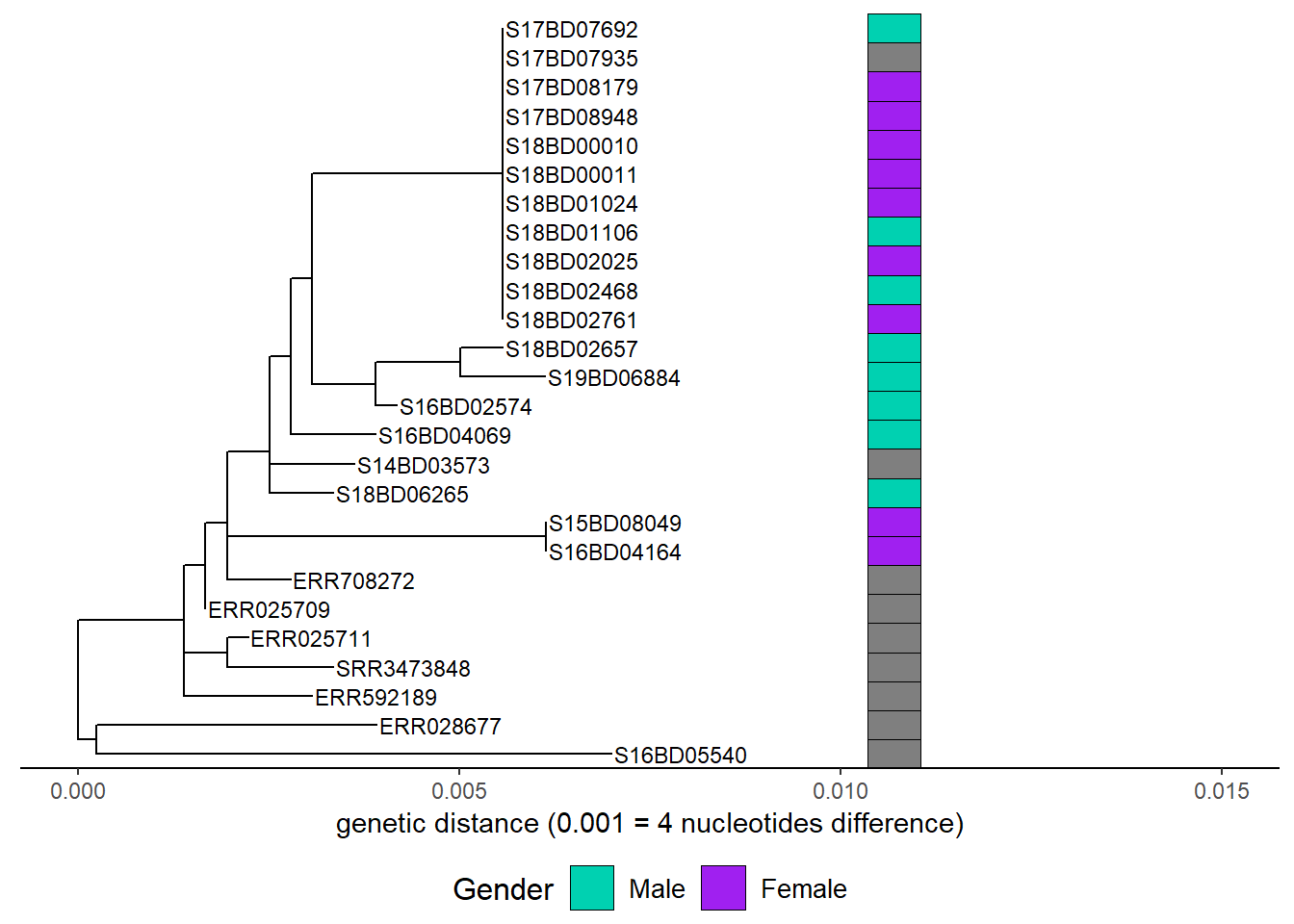
Ardından, başka bir renk şeması katmanı ekledikten sonra siprofloksasine direnç mutasyonlarını ekliyoruz:
h2 <- h1 + new_scale_fill()
h3 <- gheatmap(h2, cipR,
offset = 0.004,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_manual(name = "Ciprofloxacin resistance \n conferring mutation",
values = c("#fe9698","#ea0c92"),
breaks = c( "gyrA D87Y", "gyrA S83L"),
labels = c( "gyrA d87y", "gyrA s83l"))+
theme(legend.position = "bottom",
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.box = "vertical", legend.margin = margin())+
guides(fill = guide_legend(nrow = 2,byrow = TRUE))## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h3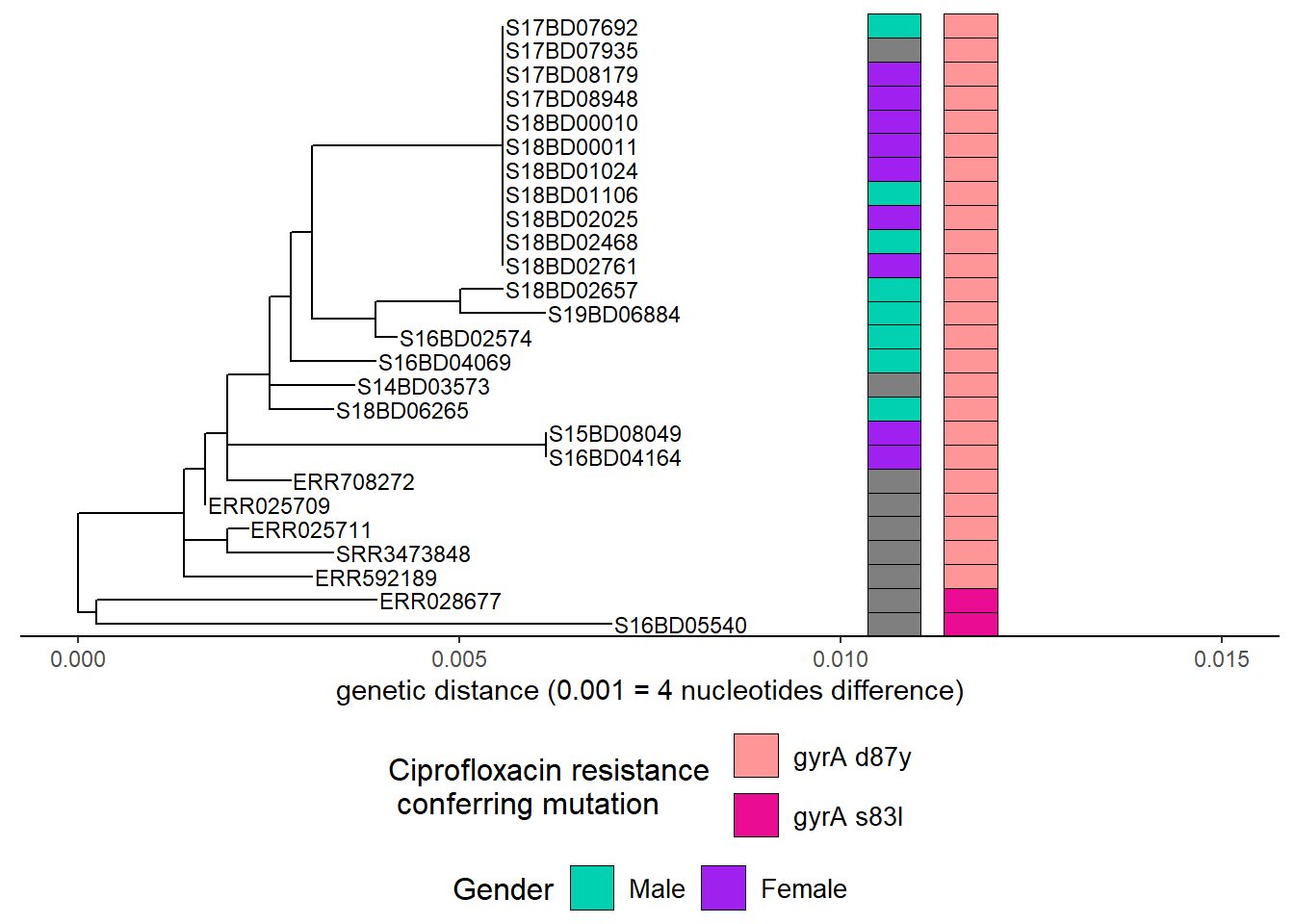
Ardından laboratuvar tarafından belirlenen minimum engelleyici konsantrasyonu (MIC) ekliyoruz:
h4 <- h3 + new_scale_fill()
h5 <- gheatmap(h4, MIC_Cip,
offset = 0.005,
width = 0.1,
color = "black",
colnames = FALSE)+
scale_fill_continuous(name = "MIC for Ciprofloxacin",
low = "yellow", high = "red",
breaks = c(0,0.50,1.00),
na.value = "white")+
guides(fill = guide_colourbar(barwidth = 5, barheight = 1))+
theme(legend.position = "bottom",
legend.title = element_text(size = 10),
legend.text = element_text(size = 8),
legend.box = "horizontal", legend.margin = margin())+
guides(shape = guide_legend(override.aes = list(size = 2)))## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## Scale for fill is already present.
## Adding another scale for fill, which will replace the existing scale.
h5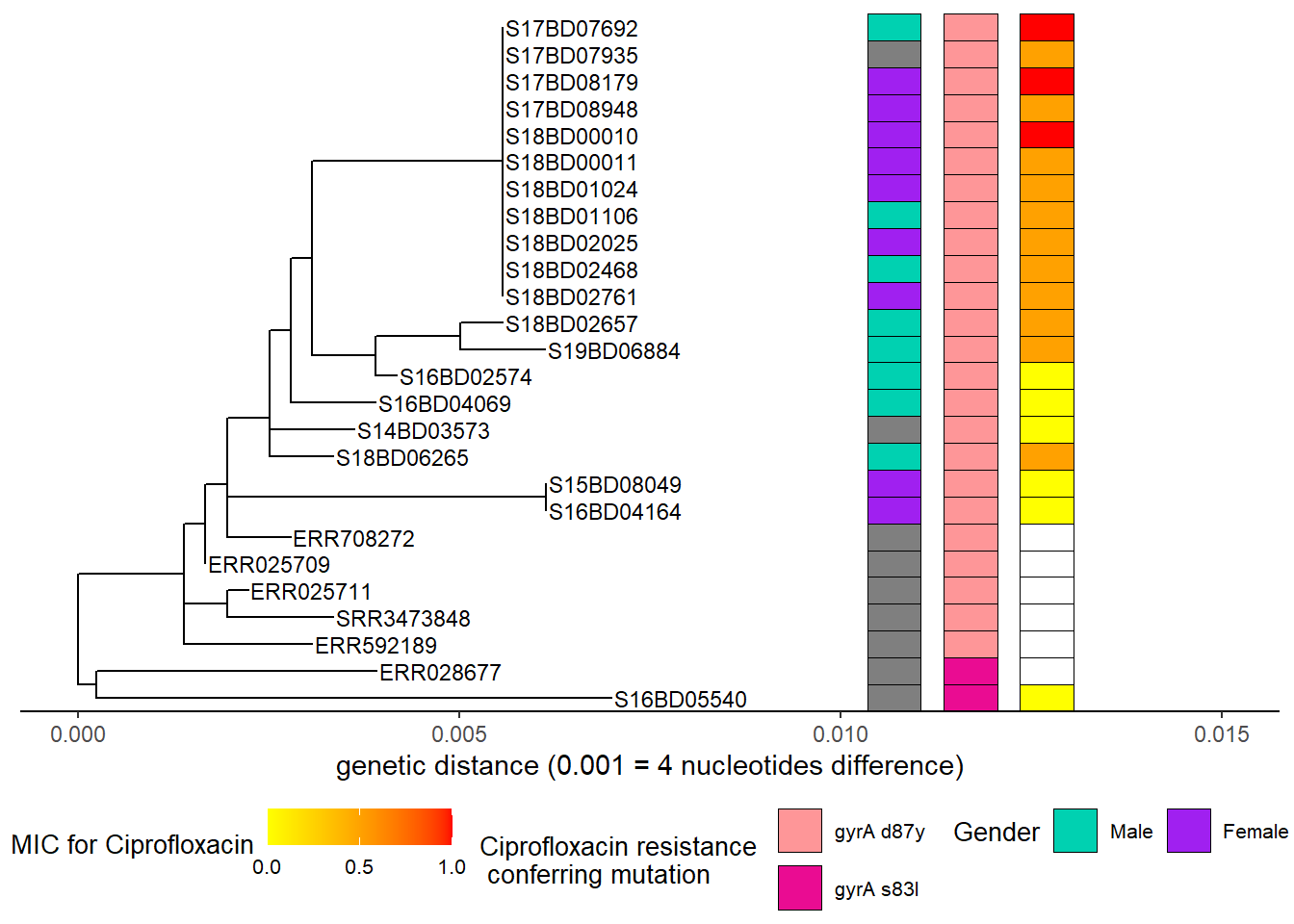
38.5 Kaynaklar
http://hydrodictyon.eeb.uconn.edu/eebedia/index.php/Ggtree# Clade_Colors https://bioconductor.riken.jp/packages/3.2/bioc/vignettes/ggtree/inst/doc/treeManipulation.html https://guangchuangyu.github.io/ggtree-book/chapter-ggtree.html https://bioconductor.riken.jp/packages/3.8/bioc/vignettes/ggtree/inst/doc/treeManipulation.html
Ea Zankari, Rosa Allesøe, Katrine G Joensen, Lina M Cavaco, Ole Lund, Frank M Aarestrup, PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens, Journal of Antimicrobial Chemotherapy, Volume 72, Issue 10, October 2017, Pages 2764–2768, https://doi.org/10.1093/jac/dkx217